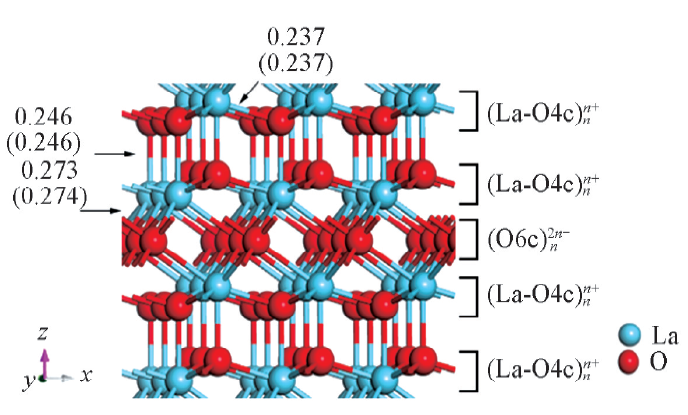
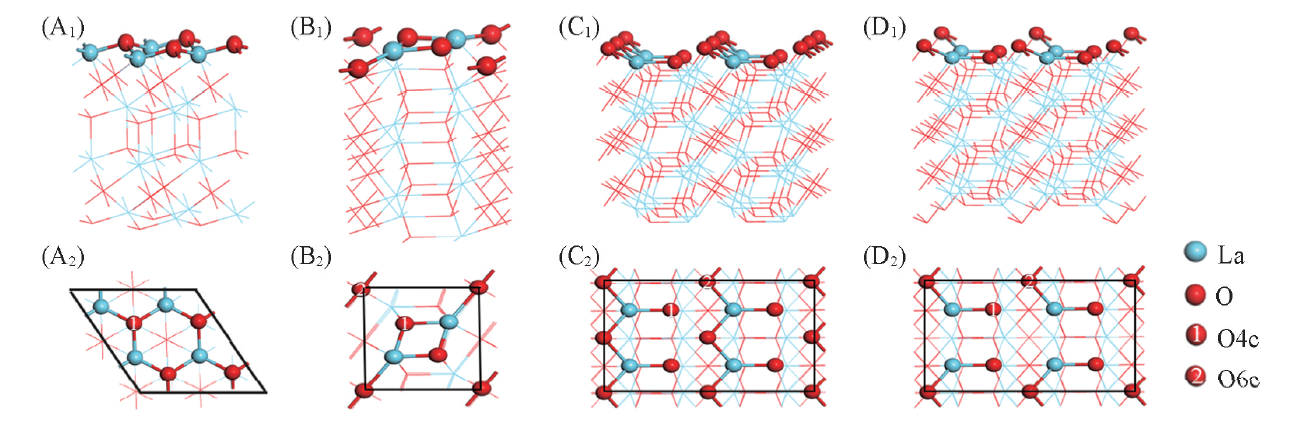
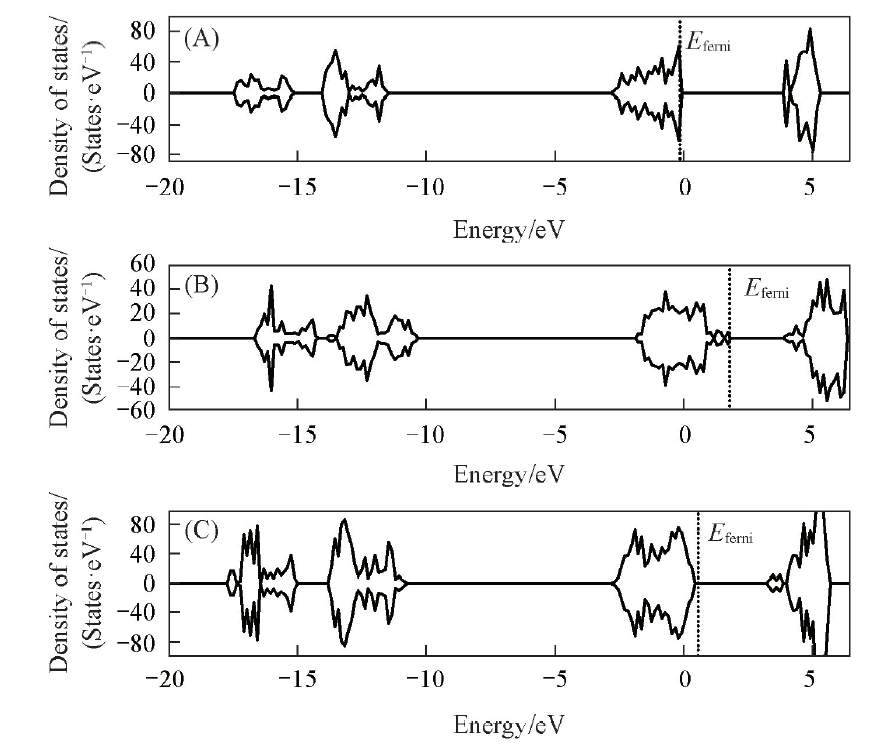
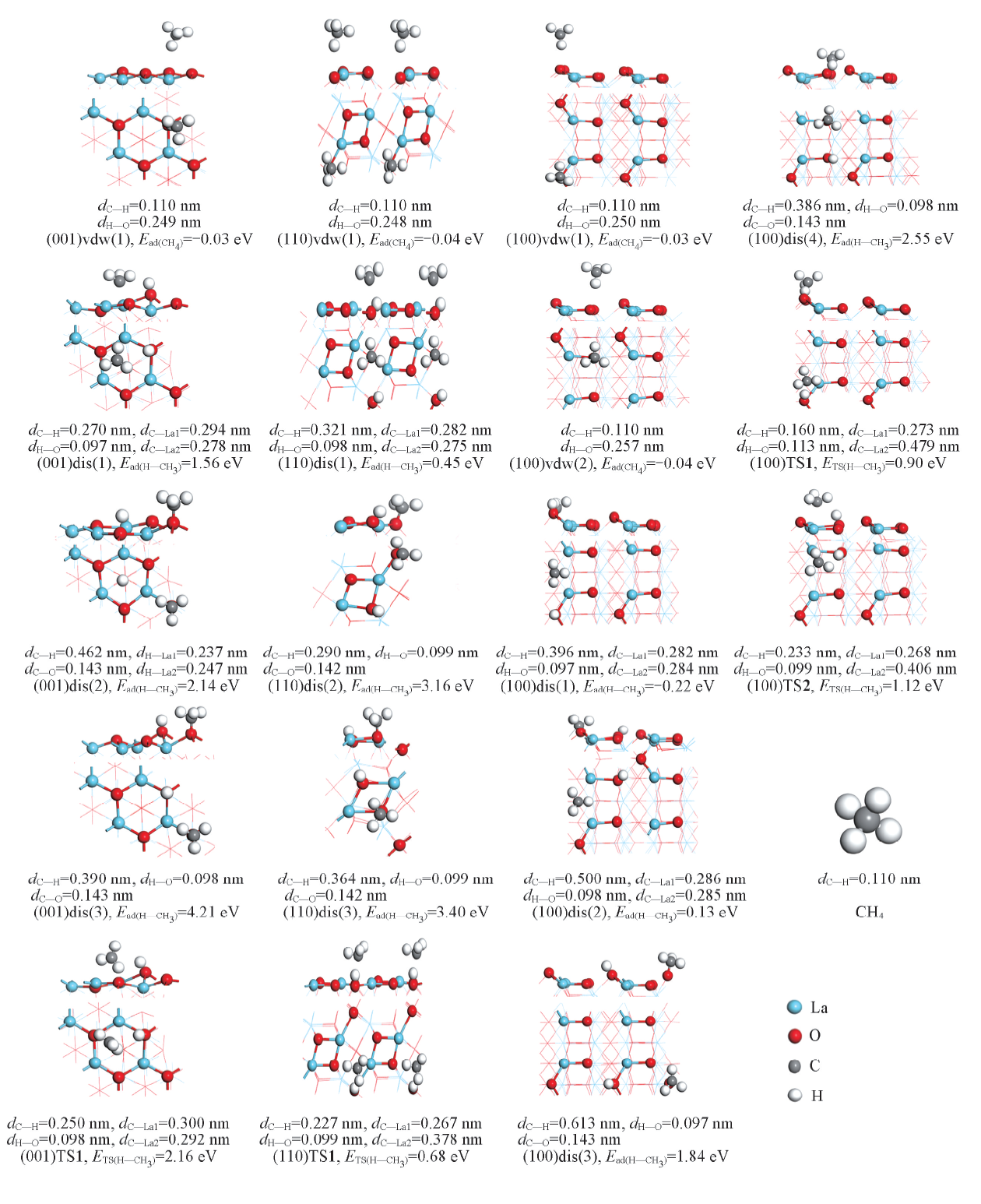
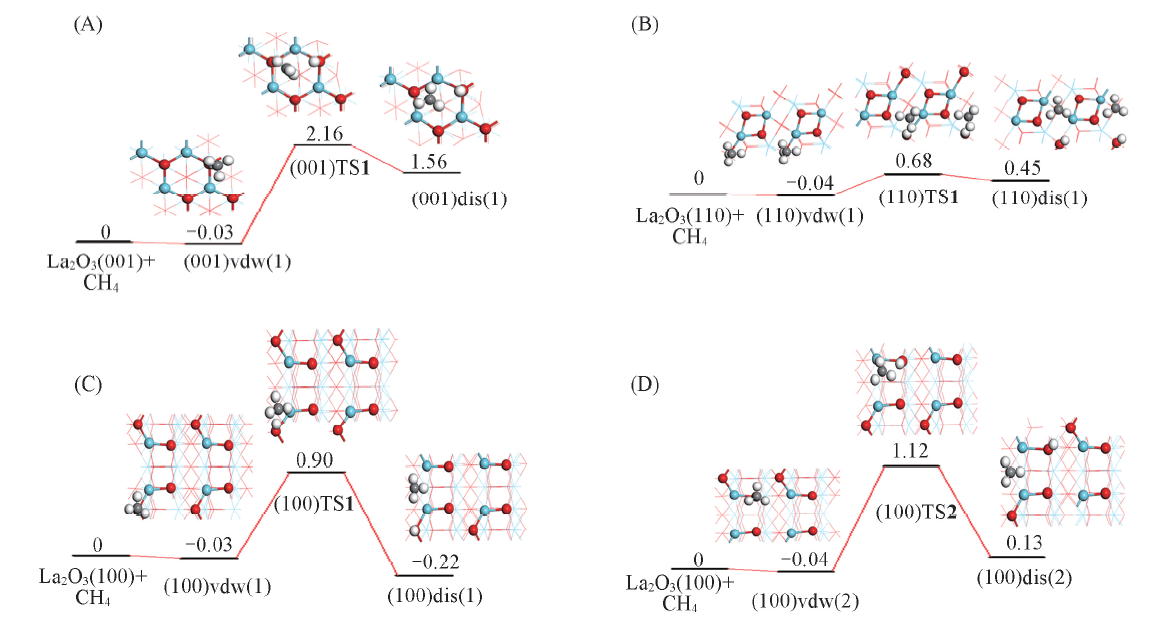
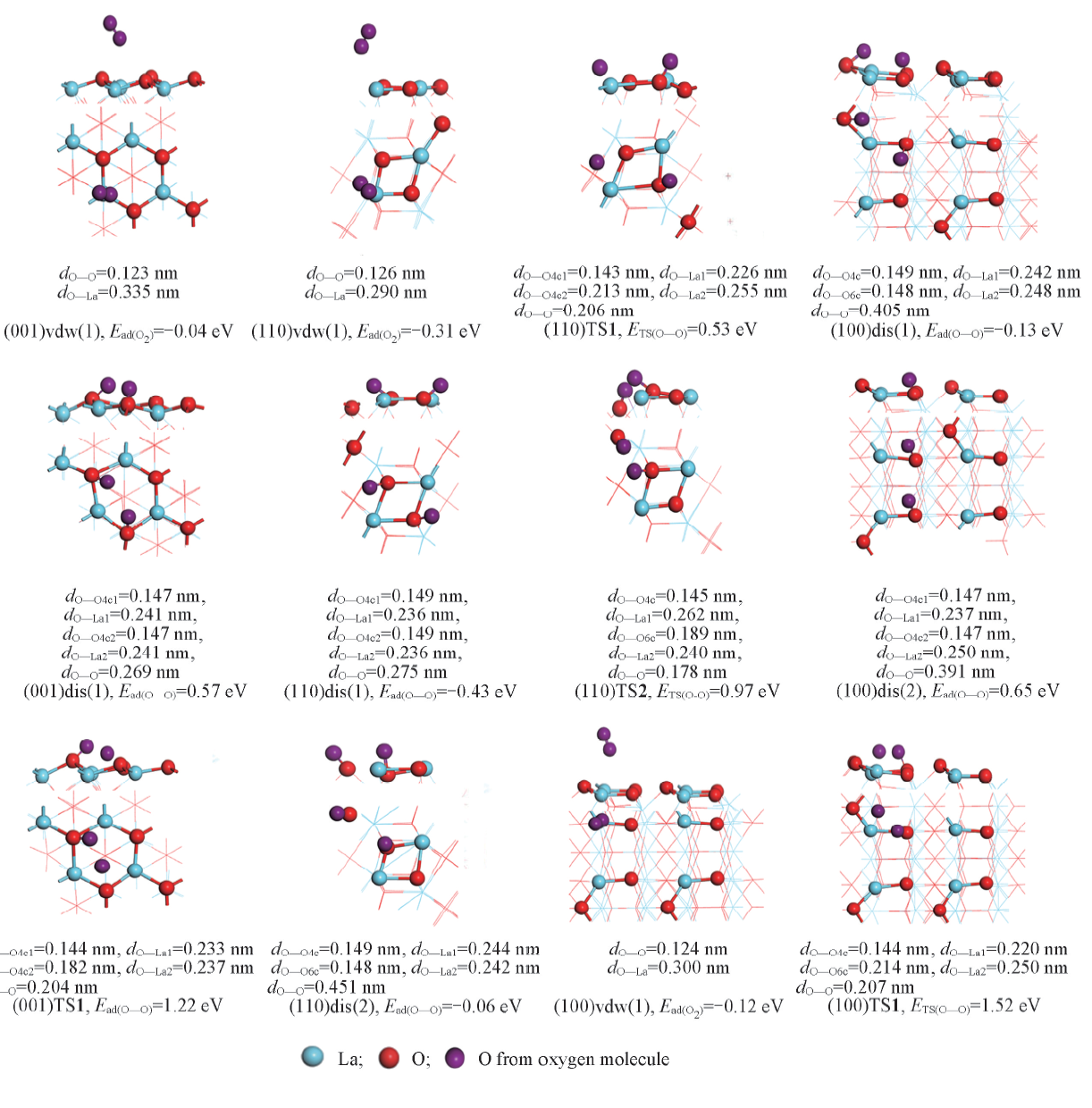
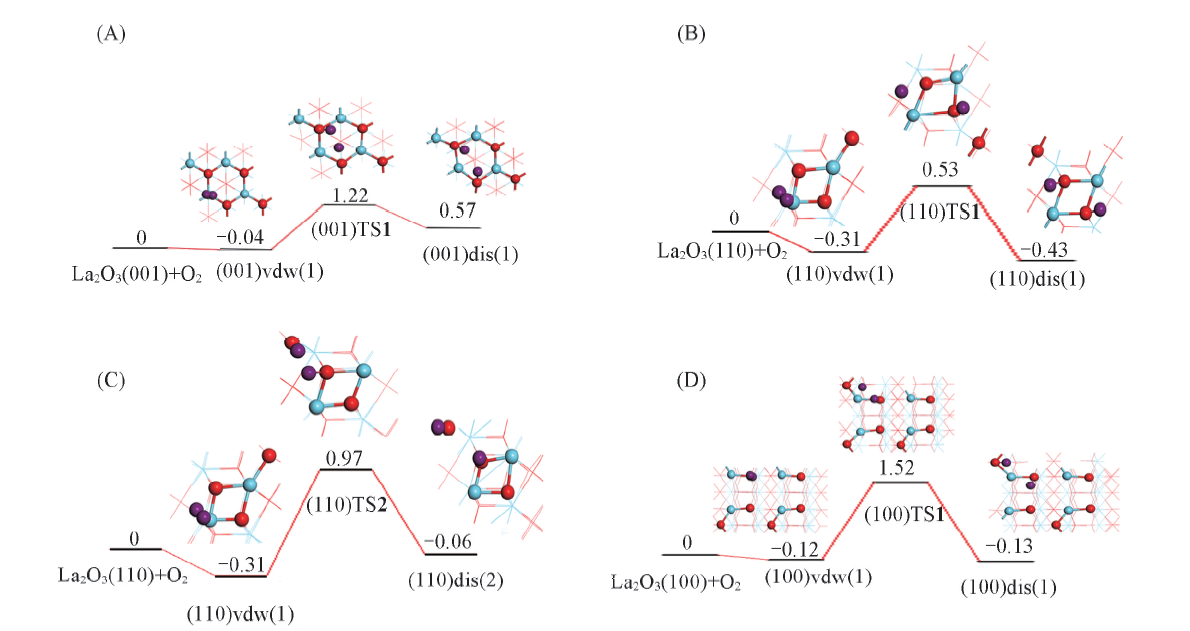
| [1] |
Schwach P., Pan X., Bao X., Chem. Rev.,2017, 117(13), 8497—8520
|
| [2] |
Kondratenko E. V., Peppel T., Seeburg D., Kondratenko V. A., Kalevaru N., Martin A., Wohlrab S., Catal. Sci. Technol.,2017, 7(2), 366—381
|
| [3] |
Wang B., Albarracin-Suazo S., Pagan-Torres Y., Nikolla E., Catal. Today,2017, 285, 147—158
|
| [4] |
Narsimhan K., Iyoki K., Dinh K., Roman-Leshkov Y., ACS Cent. Sci.,2016, 2(6), 424—429
|
| [5] |
Ravi M., Ranocchiari M., van Bokhoven J. A., Angew. Chem. Int. Ed.,2017, 56(52), 16464—16483
|
| [6] |
Zhu W., Shen M., Fan G., Yang A., Meyer J. R., Ou Y., Yin B., Fortner J., Foston M., Li Z., Zou Z., Sadtler B., ACS Appl. Nano Mater.,2018, 1(12), 6683—6691
|
| [7] |
Farrell B. L., Igenegbai V. O., Linic S., ACS Catal.,2016, 6(7), 4340—4346
|
| [8] |
Olivos-Suarez A. I., Szecsenyi A., Hensen E. J. M., Ruiz-Martinez J., Pidko E. A., Gascon J., ACS Catal.,2016, 6(5), 2965—2981
|
| [9] |
Taifan W., Baltrusaitis J., Appl. Catal. B,2016, 198, 525—547
|
| [10] |
Noon D., Seubsai A., Senkan S., ChemCatChem,2013, 5(1), 146—149
|
| [11] |
Zuo Z., Ramirez P. J., Senanayake S. D., Liu P., Rodriguez J. A., J. Am. Chem. Soc.,2016, 138(42), 13810—13813
|
| [12] |
Cui X., Li H., Wang Y., Hu Y., Hua L., Li H., Han X., Liu Q., Yang F., He L., Chen X., Li Q., Xiao J., Deng D., Bao X., Chem.,2018, 4(8), 1902—1910
|
| [13] |
Hou Y. H., Han W. C., Xia W. S., Wan H. L., ACS Catal.,2015, 5(3), 1663—1674
|
| [14] |
Greis O., J. Solid State Chem., 1980, 34(1), 39—44
|
| [15] |
Molinari M., Parker S. C., Sayle D. C., Islam M. S., J. Phys. Chem. C,2012, 116(12), 7073—7082
|
| [16] |
Kresse G., Furthmuller J., Comput. Mater. Sci.,1996, 6(1), 15—50
|
| [17] |
Kresse G., Joubert D., Phys. Rev. B: Condens. Matter Mater. Phys.,1999, 59(3), 1758—1775
|
| [18] |
Li B., Metiu H., J. Phys. Chem. C,2010, 114(28), 12234—12244
|
| [19] |
Anisimov V. I., Aryasetiawan F., Lichtenstein A. I., J. Phys.: Condens. Matter,1997, 9(4), 767—808
|
| [20] |
Perdew J. P., Zunger A., Phys. Rev. B: Condens. Matter,1981, 23(10), 5048—5079
|
| [21] |
Zhou W., Jefferson D. A., Liang W. Y., Surf. Sci.,1989, 209(3), 444—454
|
| [22] |
Islam M. S., Ilett D. J., Catal. Today,1994, 21(2/3), 417—422
|
| [23] |
Chretien S., Metiu H., J. Phys. Chem. C,2014, 118(47), 27336—27342
|
| [24] |
Palmer M. S., Neurock M., Olken M. M., J. Am. Chem. Soc.,2002, 124(28), 8452—8461
|
| [25] |
Wang S., Cong L., Zhao C., Li Y., Pang Y., Zhao Y., Li S., Sun Y., Phys. Chem. Chem. Phys.,2017, 19(39), 26799—26811
|
| [26] |
Xing B., Pang X. Y., Wang G. C., J. Catal.,2011, 282(1), 74—82
|
| [27] |
Xing B., Wang G. C., Phys. Chem. Chem. Phys.,2014, 16(6), 2621—2629
|
| [28] |
Wang J., Wang G. C., J. Phys. Chem. C,2018, 122(30), 17338—17346
|
| [29] |
Wang G. C., J. Xinyang Normal University (Nat. Sci. Ed.), 2018, 31(2), 333—338
|
|
(王贵昌. 信阳师范学院学报(自然科学版), 2018, 31(2), 333—338)
|
 ), 万惠霖
), 万惠霖