

Chem. J. Chinese Universities ›› 2014, Vol. 35 ›› Issue (5): 1000.doi: 10.7503/cjcu20140060
• Physical Chemistry • Previous Articles Next Articles
XIA Xiquan, ZHANG Hui, ZHANG Guiling*( )
)
Received:2014-01-20
Online:2014-05-10
Published:2014-04-29
Contact:
ZHANG Guiling
E-mail:1621717290@qq.com
Supported by:CLC Number:
TrendMD:
XIA Xiquan, ZHANG Hui, ZHANG Guiling. Theoretical Studies on the Structures and Spectroscopic Properties of N-Heterocyclic Carbene-pyridine-based Ruthenium Sensitizers†[J]. Chem. J. Chinese Universities, 2014, 35(5): 1000.
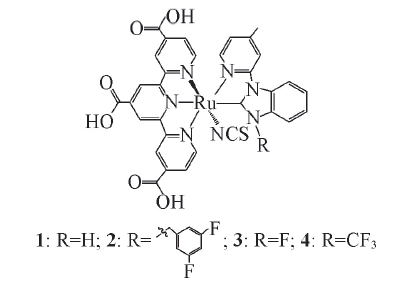
Fig.1 Schematic representation of complexes 1, 2, 3 and 4
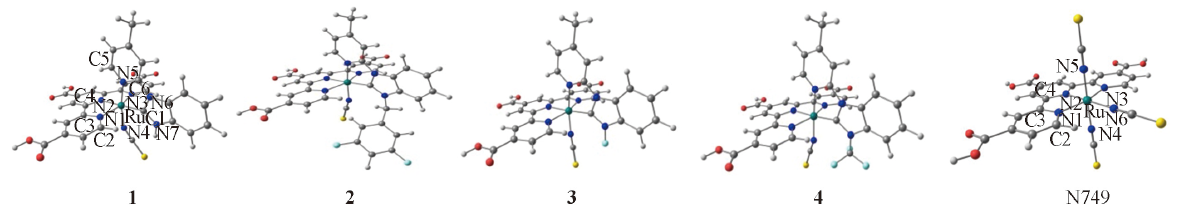
Fig.2 Optimized geometry structures of complexes 1—4 and N749 at B3LYP/LanL2DZ level of theory
| Parameter | 1 | 2 | 3 | 4 | N749 | Expt.[ |
|---|---|---|---|---|---|---|
| Ru—N1/nm | 0.2098 | 0.2106 | 0.2099 | 0.2101 | 0.2062 | 0.2090 |
| Ru—N2/nm | 0.2022 | 0.2024 | 0.2018 | 0.2026 | 0.1937 | 0.1936 |
| Ru—N3/nm | 0.2097 | 0.2095 | 0.2098 | 0.2100 | 0.2062 | |
| Ru—N4/nm | 0.2066 | 0.2040 | 0.2030 | 0.2040 | 0.2040 | 0.2032 |
| Ru—N5/nm | 0.2108 | 0.2102 | 0.2117 | 0.2101 | 0.2040 | |
| Ru—C1(N6)/nm | 0.2006 | 0.2029 | 0.2006 | 0.2019 | 0.2073 | 0.2052 |
| N1—C2/nm | 0.1361 | 0.1360 | 0.1360 | 0.1359 | 0.1358 | |
| N1—C3/nm | 0.1387 | 0.1387 | 0.1387 | 0.1386 | 0.1389 | |
| N2—C4/nm | 0.1367 | 0.1365 | 0.1367 | 0.1364 | 0.1379 | |
| N5—C5/nm | 0.1363 | 0.1363 | 0.1364 | 0.1364 | ||
| N5—C6/nm | 0.1375 | 0.1371 | 0.1372 | 0.1369 | ||
| N1—Ru—N2/(°) | 78.8 | 78.7 | 78.9 | 78.8 | 80.5 | 81.1 |
| N3—Ru—N2/(°) | 78.8 | 78.8 | 78.8 | 78.7 | 80.5 | |
| N1—Ru—N3/(°) | 157.5 | 157.6 | 157.6 | 157.4 | 161.1 | 161.6 |
| N5—Ru—C1/(°) | 77.6 | 77.9 | 77.2 | 77.6 |
Table 1 Partial optimized geometry structural parameters of complexes 1—4 and N749 in the ground states using the DFT methods and the experimental data of N749
| Parameter | 1 | 2 | 3 | 4 | N749 | Expt.[ |
|---|---|---|---|---|---|---|
| Ru—N1/nm | 0.2098 | 0.2106 | 0.2099 | 0.2101 | 0.2062 | 0.2090 |
| Ru—N2/nm | 0.2022 | 0.2024 | 0.2018 | 0.2026 | 0.1937 | 0.1936 |
| Ru—N3/nm | 0.2097 | 0.2095 | 0.2098 | 0.2100 | 0.2062 | |
| Ru—N4/nm | 0.2066 | 0.2040 | 0.2030 | 0.2040 | 0.2040 | 0.2032 |
| Ru—N5/nm | 0.2108 | 0.2102 | 0.2117 | 0.2101 | 0.2040 | |
| Ru—C1(N6)/nm | 0.2006 | 0.2029 | 0.2006 | 0.2019 | 0.2073 | 0.2052 |
| N1—C2/nm | 0.1361 | 0.1360 | 0.1360 | 0.1359 | 0.1358 | |
| N1—C3/nm | 0.1387 | 0.1387 | 0.1387 | 0.1386 | 0.1389 | |
| N2—C4/nm | 0.1367 | 0.1365 | 0.1367 | 0.1364 | 0.1379 | |
| N5—C5/nm | 0.1363 | 0.1363 | 0.1364 | 0.1364 | ||
| N5—C6/nm | 0.1375 | 0.1371 | 0.1372 | 0.1369 | ||
| N1—Ru—N2/(°) | 78.8 | 78.7 | 78.9 | 78.8 | 80.5 | 81.1 |
| N3—Ru—N2/(°) | 78.8 | 78.8 | 78.8 | 78.7 | 80.5 | |
| N1—Ru—N3/(°) | 157.5 | 157.6 | 157.6 | 157.4 | 161.1 | 161.6 |
| N5—Ru—C1/(°) | 77.6 | 77.9 | 77.2 | 77.6 |
| MO | Energy/eV | Compositions(%) | Assignment of orbital | |||
|---|---|---|---|---|---|---|
| Ru | tcterpy | NHC-py | NCS | |||
| LUMO+7 | -1.35159 | 1 | 0 | 98 | 0 | π*(NHC-py) |
| LUMO+4 | -2.12167 | 8 | 2 | 91 | 1 | π*(NHC-py) |
| LUMO+3 | -2.57447 | 1 | 99 | 1 | 0 | π*(tcterpy) |
| LUMO+2 | -2.80957 | 1 | 99 | 0 | 0 | π*(tcterpy) |
| LUMO+1 | -3.08523 | 5 | 94 | 0 | 0 | π*(tcterpy) |
| LUMO | -3.52687 | 7 | 92 | 0 | 1 | π*(tcterpy) |
| HOMO | -5.83602 | 32 | 5 | 4 | 58 | d(Ru)-π*(NCS) |
| HOMO-1 | -5.89943 | 24 | 6 | 1 | 68 | d(Ru)-π*(NCS) |
| HOMO-2 | -6.55658 | 55 | 7 | 38 | 1 | d(Ru)-π*(NHC-py) |
| HOMO-3 | -6.87142 | 36 | 13 | 9 | 41 | d(Ru)-π*(NCS) |
| HOMO-4 | -7.09863 | 48 | 15 | 2 | 35 | d(Ru)-π*(NCS) |
| HOMO-5 | -7.24748 | 3 | 3 | 95 | 1 | π(NHC-py) |
| HOMO-6 | -7.69565 | 2 | 95 | 1 | 1 | π(tcterpy) |
| HOMO-7 | -7.86817 | 18 | 4 | 76 | 1 | π(NHC-py) |
| HOMO-8 | -8.17756 | 0 | 0 | 100 | 0 | π(NHC-py) |
| HOMO-9 | -8.49675 | 2 | 96 | 0 | 1 | π(tcterpy) |
| HOMO-10 | -8.59227 | 1 | 89 | 10 | 0 | p(COOH) |
Table 2 Partial molecular orbital compositions of complex 3 in CH3CN under the TD-DFT calculations
| MO | Energy/eV | Compositions(%) | Assignment of orbital | |||
|---|---|---|---|---|---|---|
| Ru | tcterpy | NHC-py | NCS | |||
| LUMO+7 | -1.35159 | 1 | 0 | 98 | 0 | π*(NHC-py) |
| LUMO+4 | -2.12167 | 8 | 2 | 91 | 1 | π*(NHC-py) |
| LUMO+3 | -2.57447 | 1 | 99 | 1 | 0 | π*(tcterpy) |
| LUMO+2 | -2.80957 | 1 | 99 | 0 | 0 | π*(tcterpy) |
| LUMO+1 | -3.08523 | 5 | 94 | 0 | 0 | π*(tcterpy) |
| LUMO | -3.52687 | 7 | 92 | 0 | 1 | π*(tcterpy) |
| HOMO | -5.83602 | 32 | 5 | 4 | 58 | d(Ru)-π*(NCS) |
| HOMO-1 | -5.89943 | 24 | 6 | 1 | 68 | d(Ru)-π*(NCS) |
| HOMO-2 | -6.55658 | 55 | 7 | 38 | 1 | d(Ru)-π*(NHC-py) |
| HOMO-3 | -6.87142 | 36 | 13 | 9 | 41 | d(Ru)-π*(NCS) |
| HOMO-4 | -7.09863 | 48 | 15 | 2 | 35 | d(Ru)-π*(NCS) |
| HOMO-5 | -7.24748 | 3 | 3 | 95 | 1 | π(NHC-py) |
| HOMO-6 | -7.69565 | 2 | 95 | 1 | 1 | π(tcterpy) |
| HOMO-7 | -7.86817 | 18 | 4 | 76 | 1 | π(NHC-py) |
| HOMO-8 | -8.17756 | 0 | 0 | 100 | 0 | π(NHC-py) |
| HOMO-9 | -8.49675 | 2 | 96 | 0 | 1 | π(tcterpy) |
| HOMO-10 | -8.59227 | 1 | 89 | 10 | 0 | p(COOH) |
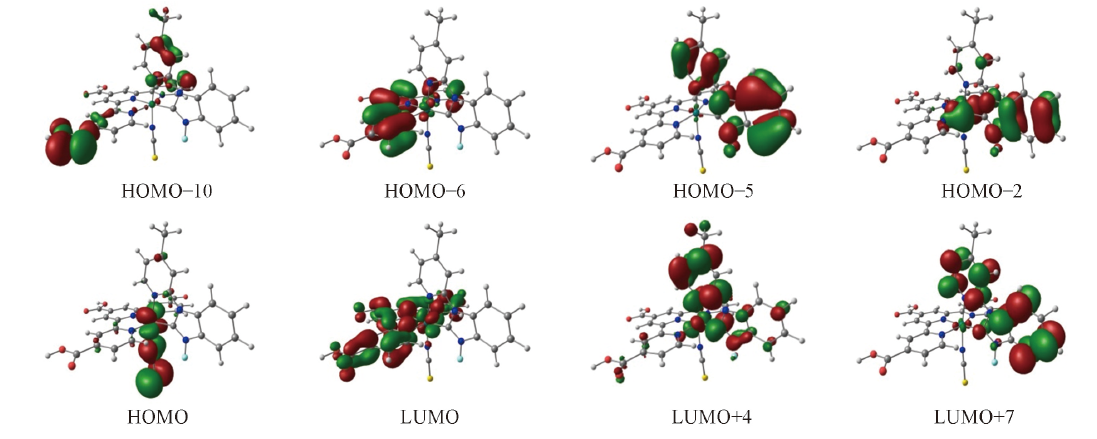
Fig.3 Electron density diagrams of the frontier molecular orbitals relevant to the absportions of complex 3 under the TD-DFT calculations
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A 1A | H→L(0.69) | 729(1.70) | 0.0329 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.61) | 610(2.03) | 0.0310 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 585(2.12) | 0.0028 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.65) | 521(2.38) | 0.0665 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.68) | 477(2.60) | 0.1015 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.63) | 370(3.35) | 0.1452 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.54) | 336(3.69) | 0.4367 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.39) | MLCT/LLCT | |||||||||
| H 1A | H-4→L+4(0.45) | 266(4.66) | 0.3790 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.28) | MLCT/LLCT | |||||||||
| 2 | A 1A | H→L(0.69) | 769(1.61) | 0.0309 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.66) | 660(1.88) | 0.0156 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 594(2.09) | 0.0017 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.69) | 546(2.27) | 0.0326 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 495(2.50) | 0.0803 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.59) | 380(3.26) | 0.1104 | MLCT/LLCT | ||||||
| G 1A | H-8→L(0.52) | 339(3.66) | 0.2986 | π(tcterpy)→π*(tcterpy) | ||||||
| H 1A | H-5→L+4(0.52) | 280(4.43) | 0.0955 | π(NHC-py)→π*(NHC-py) | ||||||
| 3 | A 1A | H→L(0.69) | 773(1.60) | 0.0259 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.68) | 683(1.82) | 0.0259 | MLCT/LLCT | ||||||
| C 1A | H-1→L+1(0.70) | 565(2.20) | 0.0266 | MLCT/LLCT | ||||||
| D 1A | H-2→L(0.69) | 557(2.23) | 0.0018 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 480(2.58) | 0.0342 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.61) | 378(3.28) | 0.1665 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.59) | 340(3.65) | 0.4973 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.31) | MLCT/LLCT | |||||||||
| H 1A | H-5→L+4(0.52) | 277(4.48) | 0.3485 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.37) | MLCT | |||||||||
| 4 | A 1A | H→L(0.70) | 777(1.60) | 0.0243 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.69) | 693(1.79) | 0.0136 | MLCT/LLCT | ||||||
| C 1A | H→L+1(0.67) | 564(2.20) | 0.0165 | MLCT/LLCT | ||||||
| D 1A | H→L+2(0.69) | 498(2.49) | 0.0511 | MLCT/LLCT | ||||||
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
| 4 | E 1A | H-3→L+1(0.61) | 382(3.24) | 0.1711 | MLCT/LLCT | |||||
| F 1A | H-6→L(0.59) | 340(3.64) | 0.5049 | π(tcterpy)→π*(tcterpy) | ||||||
| G 1A | H-5→L+4(0.58) | 275(4.50) | 0.0790 | π(NHC-py)→π*(NHC-py) | ||||||
| N749 | A 1A' | H→L+1(0.65) | 859(1.44) | 0.0409 | MLCT/LLCT | |||||
| B 1A' | H→L+2(0.57) | 686(1.80) | 0.1182 | MLCT/LLCT | 625 | |||||
| C 1A″ | H-6→L(0.66) | 513(2.42) | 0.0516 | MLCT/LLCT | 556 | |||||
| D 1A' | H-6→L+2(0.64) | 393(3.16) | 0.1586 | MLCT/LLCT | 429 | |||||
| E 1A' | H-9→L(0.64) | 334(3.72) | 0.2683 | π(tcterpy)→π*(tcterpy) | 344,330 | |||||
Table 3 Calculated absorptions of complexes 1—4 and N749 in CH3CN at TD-DFT(B3LYP) level*
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A 1A | H→L(0.69) | 729(1.70) | 0.0329 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.61) | 610(2.03) | 0.0310 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 585(2.12) | 0.0028 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.65) | 521(2.38) | 0.0665 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.68) | 477(2.60) | 0.1015 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.63) | 370(3.35) | 0.1452 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.54) | 336(3.69) | 0.4367 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.39) | MLCT/LLCT | |||||||||
| H 1A | H-4→L+4(0.45) | 266(4.66) | 0.3790 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.28) | MLCT/LLCT | |||||||||
| 2 | A 1A | H→L(0.69) | 769(1.61) | 0.0309 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.66) | 660(1.88) | 0.0156 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 594(2.09) | 0.0017 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.69) | 546(2.27) | 0.0326 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 495(2.50) | 0.0803 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.59) | 380(3.26) | 0.1104 | MLCT/LLCT | ||||||
| G 1A | H-8→L(0.52) | 339(3.66) | 0.2986 | π(tcterpy)→π*(tcterpy) | ||||||
| H 1A | H-5→L+4(0.52) | 280(4.43) | 0.0955 | π(NHC-py)→π*(NHC-py) | ||||||
| 3 | A 1A | H→L(0.69) | 773(1.60) | 0.0259 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.68) | 683(1.82) | 0.0259 | MLCT/LLCT | ||||||
| C 1A | H-1→L+1(0.70) | 565(2.20) | 0.0266 | MLCT/LLCT | ||||||
| D 1A | H-2→L(0.69) | 557(2.23) | 0.0018 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 480(2.58) | 0.0342 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.61) | 378(3.28) | 0.1665 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.59) | 340(3.65) | 0.4973 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.31) | MLCT/LLCT | |||||||||
| H 1A | H-5→L+4(0.52) | 277(4.48) | 0.3485 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.37) | MLCT | |||||||||
| 4 | A 1A | H→L(0.70) | 777(1.60) | 0.0243 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.69) | 693(1.79) | 0.0136 | MLCT/LLCT | ||||||
| C 1A | H→L+1(0.67) | 564(2.20) | 0.0165 | MLCT/LLCT | ||||||
| D 1A | H→L+2(0.69) | 498(2.49) | 0.0511 | MLCT/LLCT | ||||||
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
| 4 | E 1A | H-3→L+1(0.61) | 382(3.24) | 0.1711 | MLCT/LLCT | |||||
| F 1A | H-6→L(0.59) | 340(3.64) | 0.5049 | π(tcterpy)→π*(tcterpy) | ||||||
| G 1A | H-5→L+4(0.58) | 275(4.50) | 0.0790 | π(NHC-py)→π*(NHC-py) | ||||||
| N749 | A 1A' | H→L+1(0.65) | 859(1.44) | 0.0409 | MLCT/LLCT | |||||
| B 1A' | H→L+2(0.57) | 686(1.80) | 0.1182 | MLCT/LLCT | 625 | |||||
| C 1A″ | H-6→L(0.66) | 513(2.42) | 0.0516 | MLCT/LLCT | 556 | |||||
| D 1A' | H-6→L+2(0.64) | 393(3.16) | 0.1586 | MLCT/LLCT | 429 | |||||
| E 1A' | H-9→L(0.64) | 334(3.72) | 0.2683 | π(tcterpy)→π*(tcterpy) | 344,330 | |||||
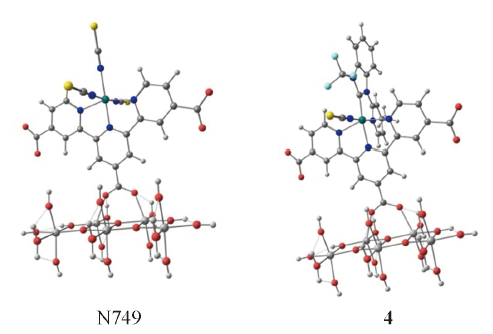
Fig.4 Optimized adsorption structure between N749, 4 and TiO2(101) surface
| [1] | O’Regan B., Grätzel M., Nature, 1991, 353, 737—740 |
| [2] | Snaith H. J., Adv. Funct. Mater., 2010, 20, 13—19 |
| [3] | Nazeeruddin M. K., Klein C., Liska P., Grätzel M., Coord. Chem. Rev., 2005, 249, 1460—1467 |
| [4] | Gao F., Wang Y., Shi D., Zhang J., Wang M., Jing X., Humphry-Baker R., Wang P., Zakeeruddin S. M., Grätzel M., J. Am. Chem. Soc., 2008, 130, 10720—10728 |
| [5] | Yum J. H., Jung I., Baik C., Ko J., Nazeeruddin M. K., Grätzel M., Energy Environ. Sci., 2009, 2, 100—102 |
| [6] | O’Regan B. C., Walley K., Juozapavicius M., Anderson A., Mater F., Ghaddar T., Zakeeruddin S. M., Klein C., Durrant J. R., J. Am. Chem. Soc., 2009, 131, 3541—3548 |
| [7] | Hagfeldt A., Boschloo G., Sun L.C., Klooand L., Pettersson H., Chem. Rev., 2010, 110, 6595—6663 |
| [8] | Zhang J., Li H. B., Wu Y., Geng Y., Duan Y. A., Liao Y., Su Z. M., Chem. J. Chinese Universities, 2011, 32(6), 1343—1348 |
| (张吉, 李海斌, 吴勇, 耿允, 段雨爱, 廖奕, 苏忠民.高等学校化学学报, 2011,32(6), 1343—1348) | |
| [9] | Nazeeruddin M. K., Kay A., Rodicio I., Humphry-Baker R., Muller E., Liska P., Vlachopoulos N., Grätzel M., J. Am. Chem. Soc., 1993, 115, 6382—6390 |
| [10] | Nazeeruddin M. K., DeAngelis F., Fantacci S., Selloni A., Viscardi G., Liska P., Ito S., Takeru B., Grätzel M., J. Am .Chem. Soc., 2005, 127, 16835—16847 |
| [11] | Nazeeruddin M.K., Pechy P., Grätzel M.,Chem. Commun., 1997, 1705—1706 |
| [12] | Nazeeruddin M. K., Pechy P., Renouard T., Zakeeruddin S. M., Humphry-Baker R., Comte P., Liska P., Cevey L., Costa E., Shklover V., Spiccia L., Deacon G. B., Bignozzi C. A., Grätzel M., J. Am. Chem. Soc., 2001, 123, 1613—1624 |
| [13] | Yu Q., Wang Y., Yi Z., Zu N., Zhang J., Zhang M., Wang P., ACS Nano, 2010, 4, 6032—6038 |
| [14] | Asghar M. I., Miettunen K., Halme J., Vahermaa P., Toivola M., Aitolaand K., Lund P., Energy Environ. Sci., 2010, 3, 418—426 |
| [15] | Wadman S.H., Kroon J. M., Bakker K., Lutz M., Spek A. L., van Klink G. P. M., van Koten G.,Chem. Commun., 2007, 1907—1909 |
| [16] | Bessho T., Yoneda E., Yum J. H., Guglielmi M., Tavernelli I., Imai H., Rothlisberger U., Nazeeruddin M. K., Grätzel M., J. Am. Chem. Soc., 2009, 131, 5930—5934 |
| [17] | Koivisto B. D., Robson K. C. D., Berlinguette C. P., Inorg. Chem., 2009, 48, 9644—9652 |
| [18] | Bomben P. G., Robson K. C. D., Koivisto B. D., Berlinguette C. P., Coord. Chem. Rev., 2012, 256, 1438—1450 |
| [19] | Brown D. G., Schauer P. A., Borau-Garcia J., Fancy B. R., Berlinguette C. P., J. Am. Chem. Soc., 2013, 135, 1692—1695 |
| [20] | Chou C.C., Wu K. L., Chi Y., Hu W. P., Yu S. J., Lee G. H., Lin C. L., Chou P. T., Angew. Chem. Int. Ed., 2011, 50, 2054—2058 |
| [21] | Chang W. C., Chen H. S., Li T. Y., Hsu N. M., Tingare Y. S., Li C. Y., Liu Y. C., Su. C. C., Li W. R., Angew. Chem. Int. Ed., 2010, 49, 8161—8164 |
| [22] | Su X., Zhang J., Wu Y., Geng Y., Su Z. M., Chem. J. Chinese Universities, 2013, 34(8), 1945—1952 |
| (苏欣, 张吉, 吴勇, 耿允, 苏忠民.高等学校化学学报, 2013,34(8), 1945—1952) | |
| [23] | Li C. M., Kan Y. H., Xu Y. Y., Duan Y. A., Su Z. M., Chem. J. Chinese Universities, 2012, 33(3), 591—597 |
| (李春敏, 阚玉和, 徐莹莹, 段雨爱, 苏忠民.高等学校化学学报, 2012,33(3), 591—597) | |
| [24] | Becke A. D., Chem. Phys., 1993, 98, 5648—5652 |
| [25] | Casida M. E., Jamorski C., Casida K. C., J. Chem. Phys., 1998, 108, 4439—4449 |
| [26] | Hay P. J., Wadt W. R., J. Chem. Phys., 1985, 82, 299—310 |
| [27] | Frisch M.J., Trucks G. W., Schlegel H. B. Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Toyota M. E. K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Peralta J. E. Jr., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J., Dapprich S., Daniels A. D., Farkas O., Foresman J., Ortiz J. V., Cioslowski J., Fox D. J., Gaussian 09, Revision A. 02, Gaussian Inc., Wallingford CT, 2009 |
| [28] | Shklover V., Nazeeruddin M. K., Grätzel M., Ovchinnikov Y. E., Appl. Organometal. Chem., 2002, 16, 635—642 |
| [29] | Fantacci S., Angelis F. D., Selloni A., J. Am. Chem. Soc., 2003, 125, 4381—4387 |
| [30] | Hagfeldt A., Grätzel M., Acc. Chem. Rev., 2000, 33, 269—277 |
| [31] | Grätzel M., Nature, 2001, 414, 338—344 |
| [1] | HE Hongrui, XIA Wensheng, ZHANG Qinghong, WAN Huilin. Density-functional Theoretical Study on the Interaction of Indium Oxyhydroxide Clusters with Carbon Dioxide and Methane [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220196. |
| [2] | WONG Honho, LU Qiuyang, SUN Mingzi, HUANG Bolong. Rational Design of Graphdiyne-based Atomic Electrocatalysts: DFT and Self-validated Machine Learning [J]. Chem. J. Chinese Universities, 2022, 43(5): 20220042. |
| [3] | LIU Yang, LI Wangchang, ZHANG Zhuxia, WANG Fang, YANG Wenjing, GUO Zhen, CUI Peng. Theoretical Exploration of Noncovalent Interactions Between Sc3C2@C80 and [12]Cycloparaphenylene Nanoring [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220457. |
| [4] | WANG Yuanyue, AN Suosuo, ZHENG Xuming, ZHAO Yanying. Spectroscopic and Theoretical Studies on 5-Mercapto-1,3,4-thiadiazole-2-thione Microsolvation Clusters [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220354. |
| [5] | CHENG Yuanyuan, XI Biying. Theoretical Study on the Fragmentation Mechanism of CH3SSCH3 Radical Cation Initiated by OH Radical [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220271. |
| [6] | ZHOU Chengsi, ZHAO Yuanjin, HAN Meichen, YANG Xia, LIU Chenguang, HE Aihua. Regulation of Silanes as External Electron Donors on Propylene/butene Sequential Polymerization [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220290. |
| [7] | MA Lijuan, GAO Shengqi, RONG Yifei, JIA Jianfeng, WU Haishun. Theoretical Investigation of Hydrogen Storage Properties of Sc, Ti, V-decorated and B/N-doped Monovacancy Graphene [J]. Chem. J. Chinese Universities, 2021, 42(9): 2842. |
| [8] | HUANG Luoyi, WENG Yueyue, HUANG Xuhui, WANG Chaojie. Theoretical Study on the Structures and Properties of Flavonoids in Plantain [J]. Chem. J. Chinese Universities, 2021, 42(9): 2752. |
| [9] | ZHONG Shengguang, XIA Wensheng, ZHANG Qinghong, WAN Huilin. Theoretical Study on Direct Conversion of CH4 and CO2 into Acetic Acid over MCu2Ox(M = Cu2+, Ce4+, Zr4+) Clusters [J]. Chem. J. Chinese Universities, 2021, 42(9): 2878. |
| [10] | ZHENG Ruoxin, ZHANG Igor Ying, XU Xin. Development and Benchmark of Lower Scaling Doubly Hybrid Density Functional XYG3 [J]. Chem. J. Chinese Universities, 2021, 42(7): 2210. |
| [11] | YING Fuming, JI Chenru, SU Peifeng, WU Wei. λ-DFCAS: A Hybrid Density Functional Complete Active Space Self Consistent Field Method [J]. Chem. J. Chinese Universities, 2021, 42(7): 2218. |
| [12] | WANG Jian, ZHANG Hongxing. Theoretical Study on the Structural-photophysical Relationships of Tetra-Pt Phosphorescent Emitters [J]. Chem. J. Chinese Universities, 2021, 42(7): 2245. |
| [13] | HU Wei, LIU Xiaofeng, LI Zhenyu, YANG Jinlong. Surface and Size Effects of Nitrogen-vacancy Centers in Diamond Nanowires [J]. Chem. J. Chinese Universities, 2021, 42(7): 2178. |
| [14] | YANG Yiying, ZHU Rongxiu, ZHANG Dongju, LIU Chengbu. Theoretical Study on Gold-catalyzed Cyclization of Alkynyl Benzodioxin to 8-Hydroxy-isocoumarin [J]. Chem. J. Chinese Universities, 2021, 42(7): 2299. |
| [15] | LIU Yang, LI Qingbo, SUN Jie, ZHAO Xian. Direct Synthesis of Graphene on AlN Substrates via Ga Remote Catalyzation [J]. Chem. J. Chinese Universities, 2021, 42(7): 2271. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||