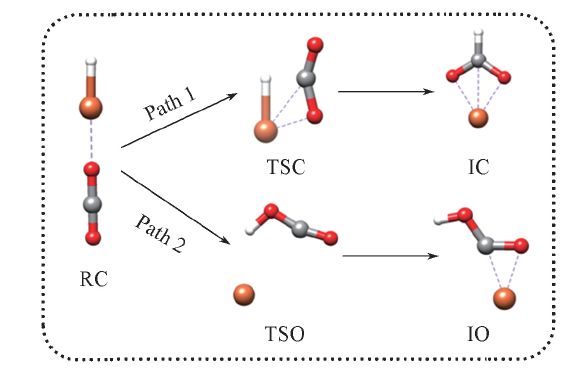
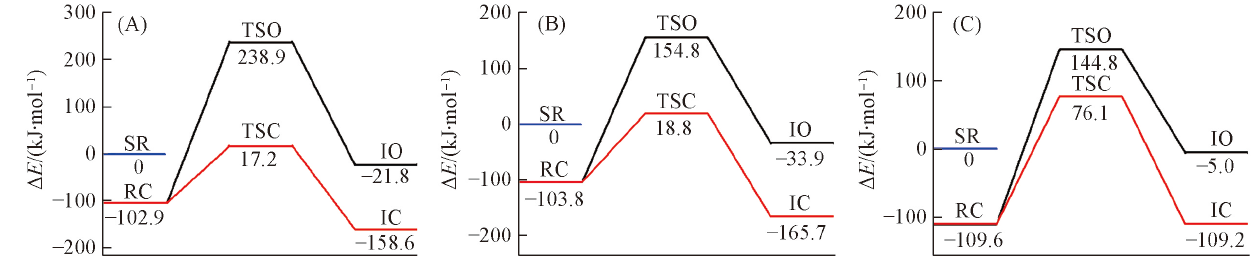
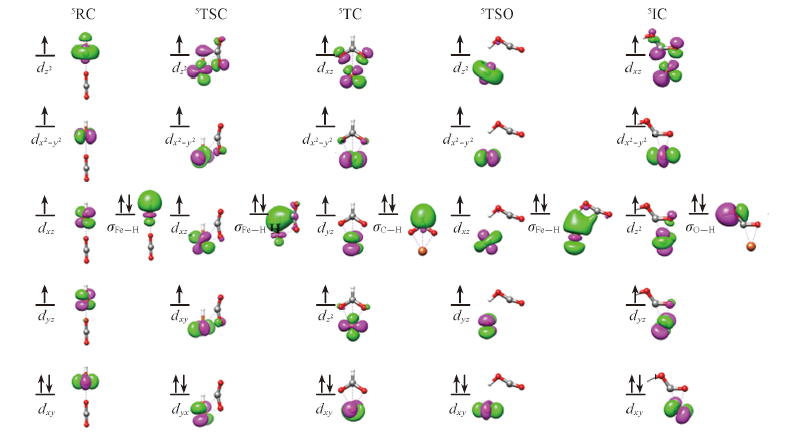
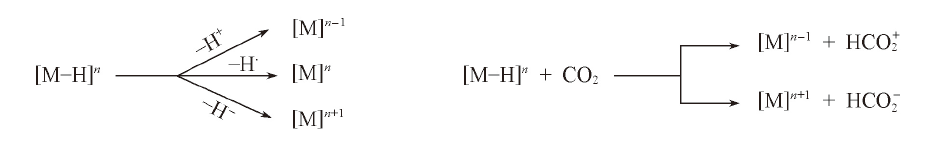| [1] |
Harrison J. F., Chem. Rev., 2000, 100, 679—716
|
| [2] |
Yin X., Moss J. R., Coord. Chem. Rev., 1999, 181, 27—59
|
| [3] |
Musashi Y., Sakaki S., J. Am. Chem. Soc., 2000, 122, 3867—3877
|
| [4] |
Darensbourg D. J., Inorg. Chem ., 2010, 49, 10765—10780
|
| [5] |
Roberts J. A., Appel A. M., DuBois D. L., Bullock R. M., J. Am. Chem. Soc., 2011, 133, 14604—14613
|
| [6] |
Tate B. K., Wyss C. M., Bacsa J., Kluge K., Gelbaum L., Sadighi J. P., Chem. Sci., 2013, 4, 3068—3074
|
| [7] |
Jiang Y. F., Blacque O., Fox T., Berke H., J. Am. Chem. Soc., 2013, 135, 7751—7160
|
| [8] |
Alapati S. V., Karl J. J., Sholl D. S., Phys. Chem. Chem. Phys., 2007, 9, 1438—1452
|
| [9] |
Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas ., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J., Gaussian 09, Gaussian Inc.,Wallingford CT, 2009
|
| [10] |
Fukui K., J. Phys. Chem., 1970, 74, 4161—4163
|
| [11] |
Neese F., ORCA-an ab initio, Density Functional and Semiempirical Program Package 2010, Version 2.8, Bonn University,Bonn, 2010
|
| [12] |
Neese F., J. Am. Chem. Soc., 2006, 128, 10213—10222
|
| [13] |
Sun X. L., Huang X. R., Li J. L., Huo R. P., Sun C. C., J. Phys. Chem. A, 2012, 116, 1475—1485
|
| [14] |
Sun X. L., Geng C. Y., Huo R. P., Ryde U., Bu Y. X., Li J. L., J. Phys. Chem. B, 2014, 118, 1493—1500
|
| [15] |
Sun X. H., Sun X. L., Geng C. Y., Zhao H. T., Li J. L., J. Phys. Chem. A, 2014, 118, 7146—7158
|
| [16] |
Huo R. P., Zhang X., Huang X. R., Li J. L., Sun C. C. J. Mol.Model., 2013, 19, 1009—1018
|
| [17] |
Sun X. L., Li J. L., Huang X. R., Sun C. C., Acta Chim. Sinica, 2012, 70, 1245—1249
|
|
(孙小丽, 李吉来, 黄旭日, 孙家锺. 化学学报, 2012, 70, 1245—1249)
|
| [18] |
Huo R. P., Zhang X., Huang X. R., Li J. L., Sun C. C., J. Phys. Chem. A, 2011, 115, 3576—3582
|
| [19] |
Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem., 2004, 25, 1605—1612
|
| [20] |
Liu S., Geng Z., Wang Y., Yan Y., J. Phys. Chem. A, 2012, 116, 4560—4568
|
| [21] |
Zhang Q., Bowers M. T., J. Phys. Chem. A, 2004, 108, 9755—9761
|
 )
)