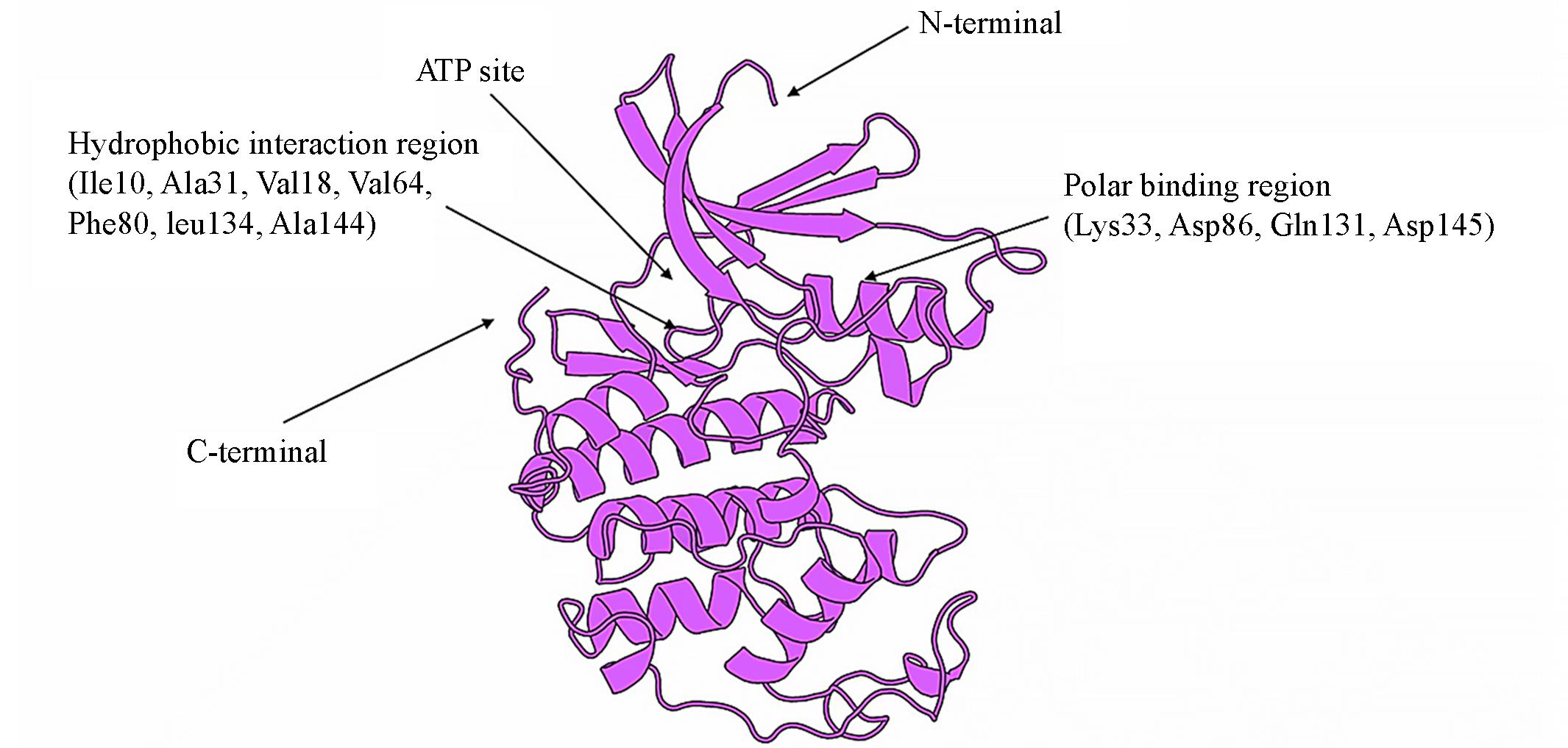
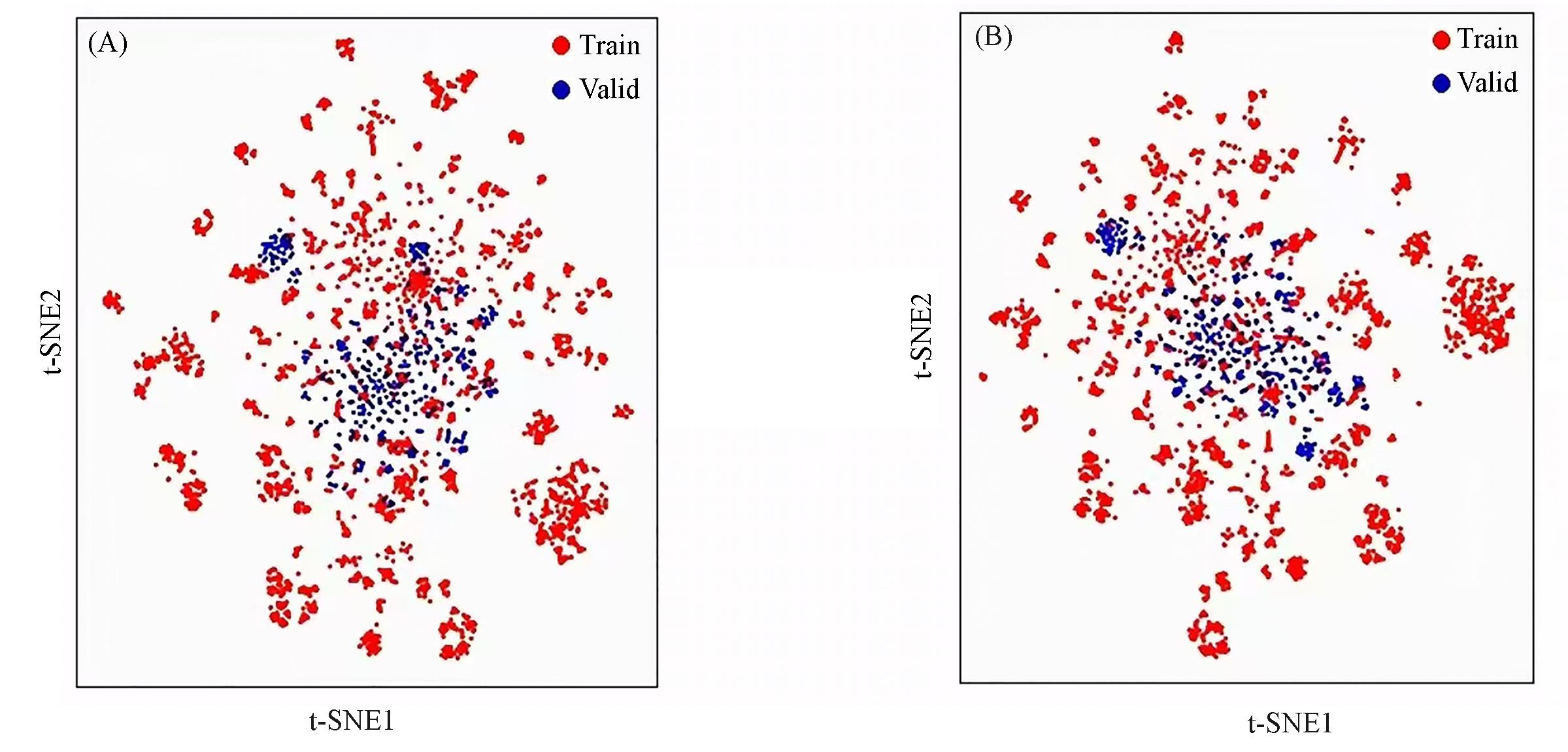
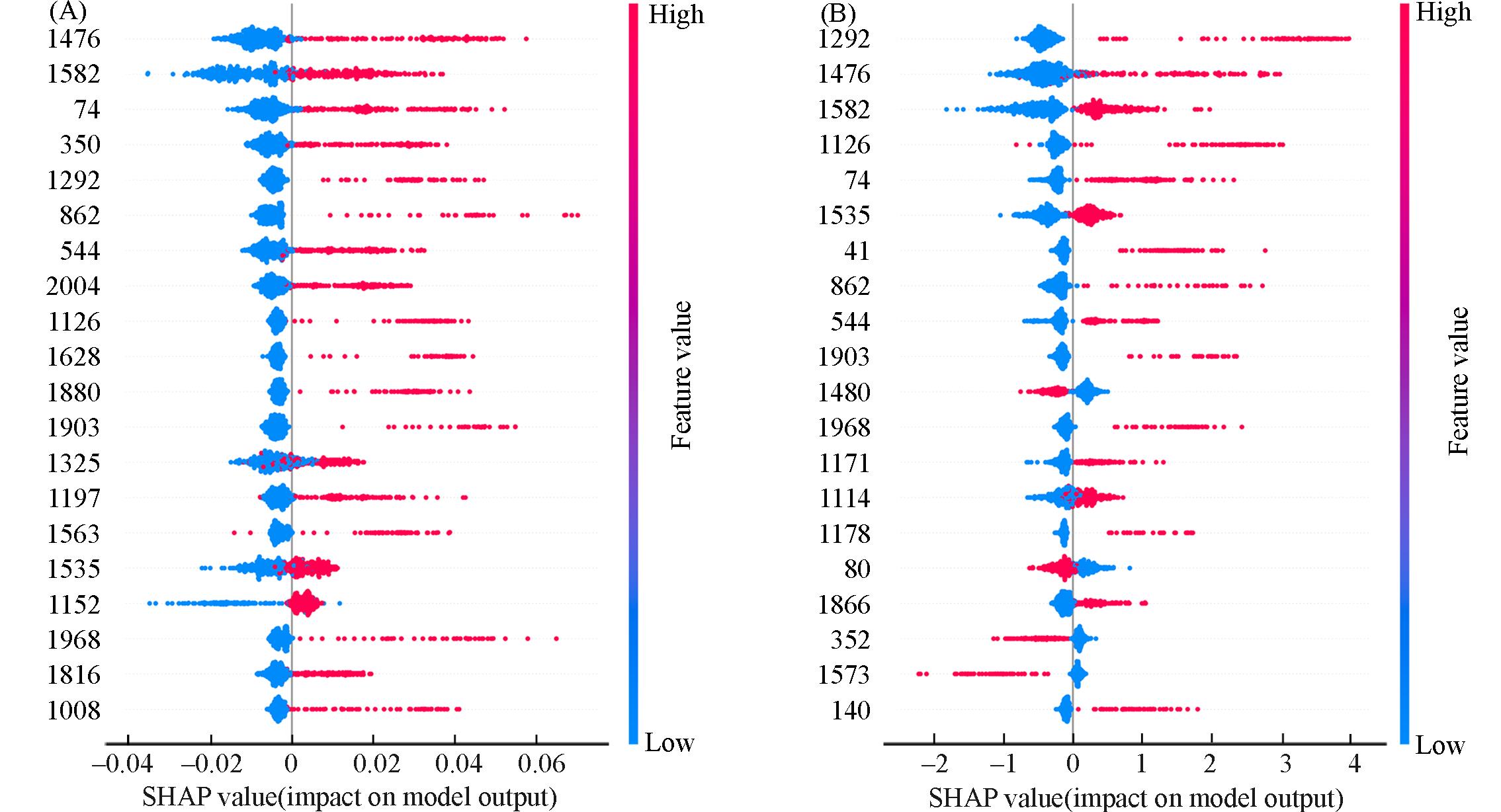
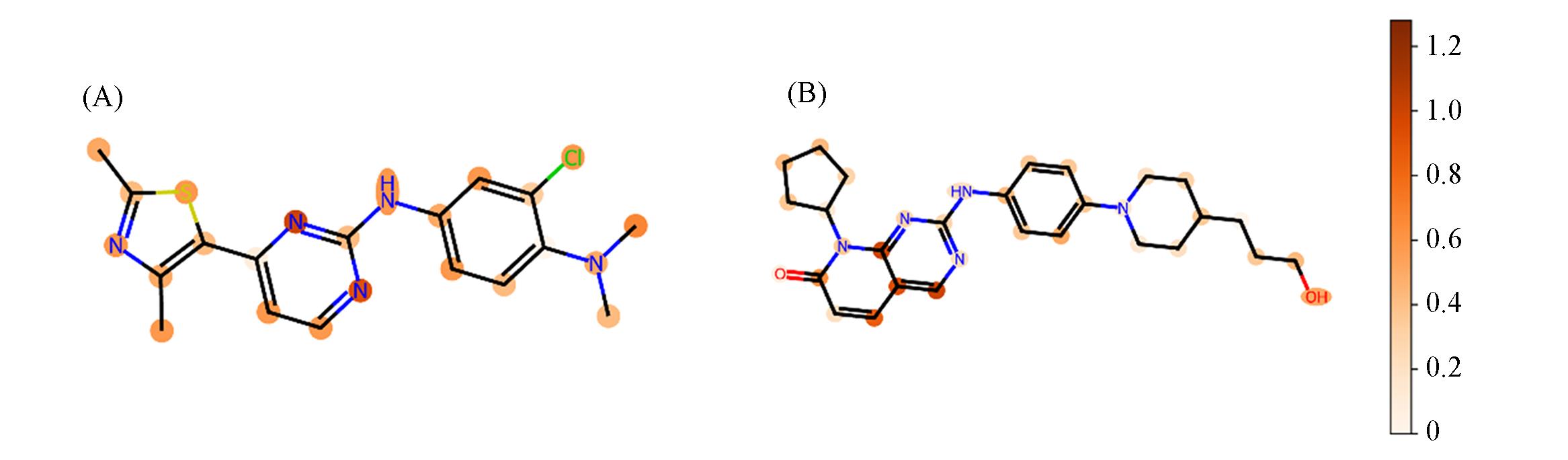
| 1 |
Swaffer M. P., Jones A. W., Flynn H. R., Snijders A. P., Nurse P., Cell, 2016, 167(7), 1750—1761
|
| 2 |
Arellano M., Moreno S., Int. J. Biochem. Cell. Biol., 1997, 29(4), 559—573
|
| 3 |
Matsuura I., Denissova N. G., Wang G., He D., Long J., Liu F., Nature, 2004, 430(6996), 226—231
|
| 4 |
Matsumoto Y., Hayashi K., Nishida E., Curr. Biol., 1999, 9(8), 429—432
|
| 5 |
Pagano M., Pepperkok R., Lukas J., Baldin V., Ansorge W., Bartek J., Draetta G., J. Cell. Biol., 1993, 121(1), 101—111
|
| 6 |
Hu S., Danilov A. V., Godek K., Orr B., Tafe L. J., Rodriguez⁃Canales J., Behrens C., Mino B., Moran C. A., Memoli V. A., Mustachio L. M., Cancer Res., 2015, 75(10), 2029—2038
|
| 7 |
Tetsu O., Mccormick F., Cancer Cell, 2003, 3(3), 233—245
|
| 8 |
Faber A. C., Chiles T. C., Cell Cycle, 2007, 6(23), 2982—2989
|
| 9 |
Karst A. M., Jones P. M., Vena N., Ligon A. H., Liu J. F., Hirsch M. S., Etemadmoghadam D., Bowtell D. D., Drapkin R., Cancer Res., 2014, 74(4), 1141—1152
|
| 10 |
Keck J. M., Summers M. K., Tedesco D., Ekholm⁃Reed S., Chuang L. C., Jackson P. K., Reed S. I., J. Cell. Biol., 2007, 178(3), 371—385
|
| 11 |
Sonntag R., Giebeler N., Nevzorova Y. A., Bangen J. M., Fahrenkamp D., Lambertz D., Haas U., Hu W., Gassler N., Cubero F. J., Müller⁃Newen G., Proc. Natl. Acad. Sci., 2018, 115(37), 9282—9287
|
| 12 |
Tadesse S., Caldon E. C., Tilley W., Wang S., J. Med. Chem., 2019, 62(9), 4233—4251
|
| 13 |
Tadesse S., Anshabo A. T., Portman N., Lim E., Tilley W., Caldon C. E., Wang S., Drug Discov. Today, 2020, 25(2), 406—413
|
| 14 |
Beale G., Haagensen E. J., Thomas H. D., Wang L. Z., Revill C. H., Payne S. L., Golding B. T., Hardcastle I. R., Newell D. R., Griffin R. J., Cano C., Br. J. Cancer, 2016, 115(6), 682—690
|
| 15 |
Cheng C. K., Gustafson W. C., Charron E., Houseman B. T., Zunder E., Goga A., Gray N. S., Pollok B., Oakes S. A., James C. D., Shokat K. M., Proc. Natl. Acad. Sci., 2012, 109(31), 12722—12727
|
| 16 |
Tripathi S. K., Muttineni R., Singh S. K., J. Theor. Biol., 2013, 334, 87—100
|
| 17 |
Rogers D., Hahn M., J. Chem. Inf. Model, 2010, 50(5), 742—754
|
| 18 |
Belkina A. C., Ciccolella C. O., Anno R., Halpert R., Spidlen J., Snyder⁃Cappione J. E., Nat. Commun., 2019, 10(1), 5415
|
| 19 |
Kobak D., Berens P., Nat. Commun., 2019, 10(1), 5416
|
| 20 |
Su X., Bai M., PLoS One, 2020, 15(8), e0238000
|
| 21 |
Sheridan R. P., Wang W. M., Liaw A., Ma J., Gifford E. M., J. Chem. Inf. Model., 2016, 56(12), 2353—2360
|
| 22 |
Lundberg S. M., Erion G., Chen H., DeGrave A., Prutkin J. M., Nair B., Katz R., Himmelfarb J., Bansal N., Lee S. I., Nat. Mach. Intell., 2020, 2(1), 56—67
|
| 23 |
Rodríguez⁃Pérez R., Bajorath J., J. Med. Chem., 2020, 63(16), 8761—8777
|
| 24 |
Trott O., Olson A. J., J. Comput. Chem., 2010, 31(2), 455—461
|
| 25 |
Daina A., Michielin O., Zoete V., Sci. Rep., 2017, 7(1), 42717
|
| 26 |
Xiong G., Wu Z., Yi J., Fu L., Yang Z., Hsieh C., Yin M., Zeng X., Wu C., Lu A., Chen X., Nucleic Acids Res., 2021, 49(W1), W5—W14
|
| 27 |
Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams⁃Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J., Gaussian 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016
|
| 28 |
Tirado⁃Rives J., Jorgensen W. L., J. Chem. Theory. Comput., 2008, 4(2), 297—306
|
| 29 |
Olsson M. H., Søndergaard C. R., Rostkowski M., Jensen J. H., J. Chem. Theory. Comput., 2011, 7(2), 525—537
|
| 30 |
Case D. A., Cheatham III T. E., Darden T., Gohlke H., Luo R., Merz Jr K. M., Onufriev A., Simmerling C., Wang B., Woods R. J., J. Comput. Chem., 2005, 26(16), 1668—1688
|
| 31 |
He X., Man V. H., Yang W., Lee T. S., Wang J., J. Chem. Phys., 2020, 153(11), 114502
|
| 32 |
Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys., 1983, 79(2), 926—935
|
| 33 |
Hess B., Bekker H., Berendsen H. J., Fraaije J. G., J. Comput. Chem., 1997, 18(12), 1463—1472
|
| 34 |
Darden T. A., York D. M., Pedersen L. G., J. Chem. Phys., 1993, 98(12), 10089—10092
|
| 35 |
Kutzner C., Páll S., Fechner M., Esztermann A., de Groot B. L., Grubmüller H., J. Comput. Chem., 2019, 40(27), 2418—2431
|
| 36 |
Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 1996, 14(1), 33—38
|
| 37 |
DeLano W. L, Protein Crystallogr., 2002, 40(1), 82—92
|
| 38 |
Dai Y., Wang Q., Zhang X., Jia S., Zheng H., Feng D., Yu P., Eur. J. Med. Chem., 2010, 45(12), 5612—5620
|
| 39 |
Sun H., Duan L., Chen F., Liu H., Wang Z., Pan P., Zhu F., Zhang J. Z., Hou T., Phys. Chem. Chem. Phys., 2018, 20(21), 14450—14460
|
| 40 |
Lundberg S. M., Erion G., Chen H., DeGrave A., Prutkin J. M., Nair B., Katz R., Himmelfarb J., Bansal N., Lee S. I., Nat. Mach. Intell., 2020, 2(1), 56—67
|
| 41 |
Rodríguez⁃Pérez R., Bajorath J., J. Med. Chem., 2020, 63(16), 8761—8777
|
 )
)