高等学校化学学报 ›› 2021, Vol. 42 ›› Issue (9): 2878.doi: 10.7503/cjcu20210282
钟声广, 夏文生( ), 张庆红, 万惠霖
), 张庆红, 万惠霖
ZHONG Shengguang, XIA Wensheng(), ZHANG Qinghong, WAN Huilin
摘要:
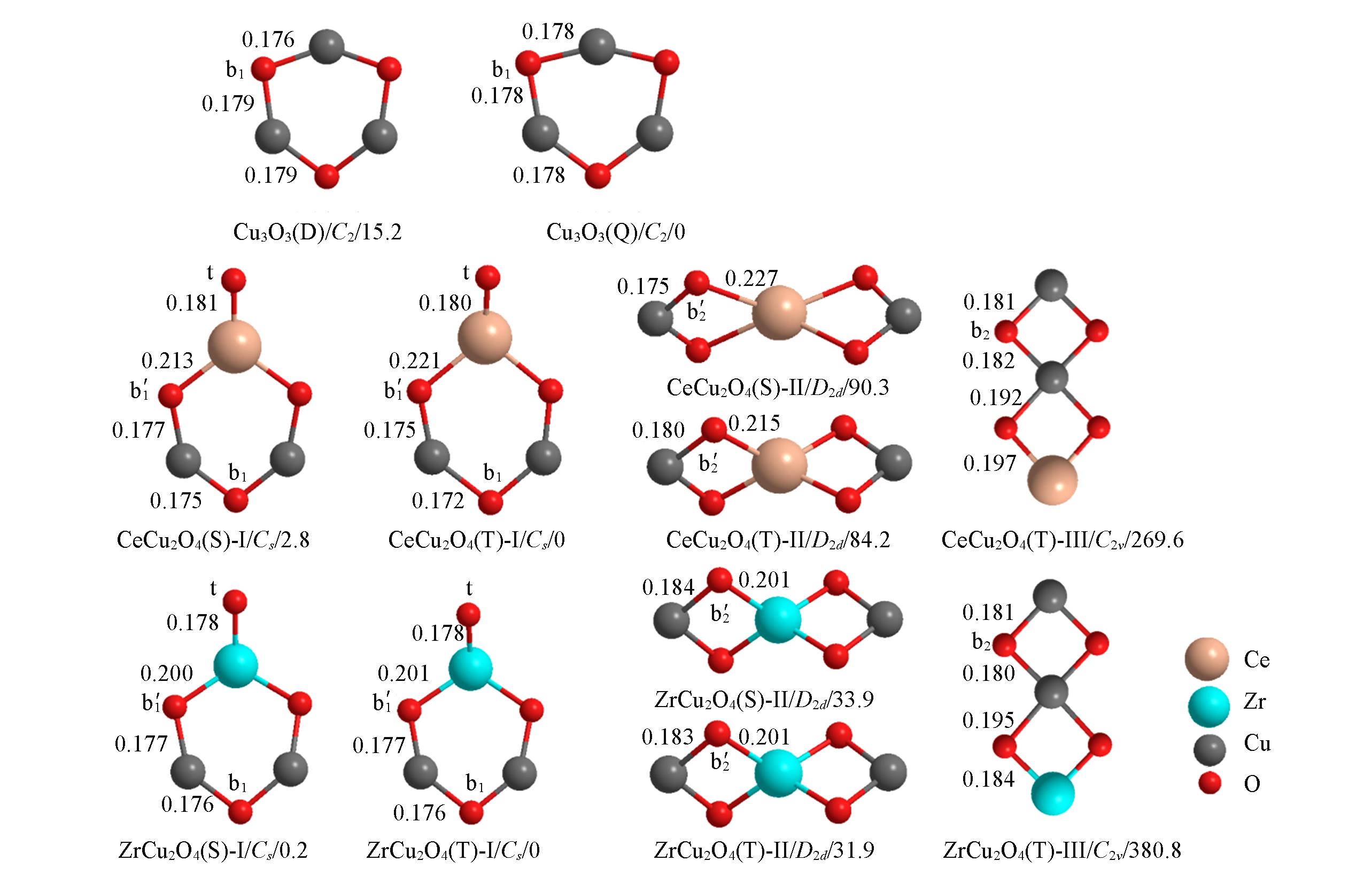
采用密度泛函理论(DFT)方法研究了电中性团簇MCu2Ox(M=Cu2+, Ce4+, Zr4+; x=3, 4)的特性及其对甲烷和二氧化碳直接合成乙酸反应的影响. 结果表明, 团簇催化的反应由甲烷C—H活化、 二氧化碳插入引起C-C偶联、 CH3COO转向和氢迁移4步构成. 前两步为关键步骤, C—H和C-C各自与团簇活性位点间形成四中心结构并推动反应进行. 电子自甲烷流出到团簇, 再流入二氧化碳, 使甲烷的C—H和二氧化碳的C=O得以活化, 继而驱动C-C偶联. Ce, Zr引入至氧化铜团簇中后, 团簇由原有的六元环结构衍变为六元环Ⅰ、 掺杂原子分别位于中心和端末的双四元环Ⅱ和Ⅲ 3种结构. 团簇结构和电子自旋均会影响反应的进行. 低自旋团簇有利于甲烷 C—H活化, 而高自旋团簇则有利于C-C偶联; 在3种掺杂团簇结构中, 处于三重态的结构Ⅲ团簇可以较好地兼顾C—H活化和C-C偶联. 通过比较相同结构发现, Ce, Zr掺杂调变了氧化铜团簇活性位点的局域电荷, 虽使其对甲烷C—H活化的能力略有下降, 但却显著降低了C-C偶联反应的活化自由能垒, 从而促进了反应的进行. 掺杂原子Zr的助剂作用比Ce要大.
中图分类号:
TrendMD: