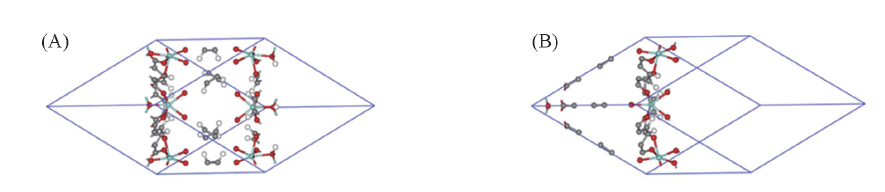
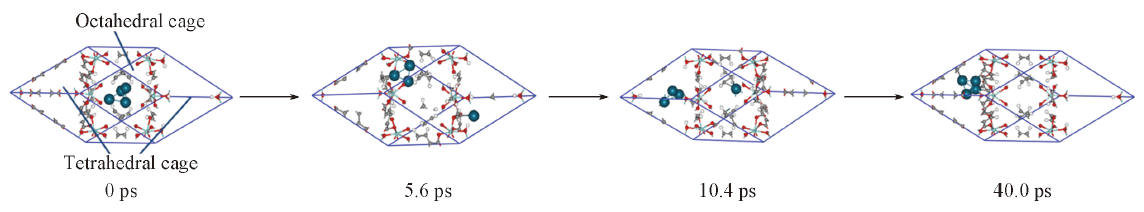
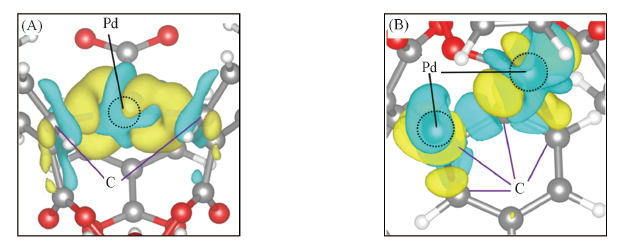
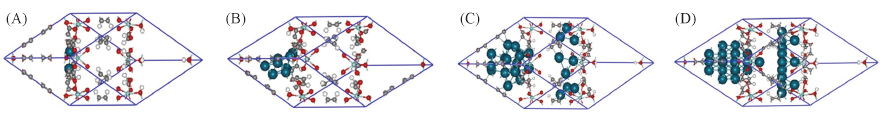
| [1] |
Zhou H. C., Kitagawa S., Chem. Soc. Rev., 2014, 43, 5415—5418
|
| [2] |
Zhu Q. L., Xu Q., Chem. Soc. Rev., 2014, 43, 5468—5512
|
| [3] |
Wu J., Zhao B. W., Huang C., Chem. J. Chinese Universities,2016, 37(6), 1069—1074
|
|
(吴娟, 赵博文, 黄超.高等学校化学学报, 2016, 37(6), 1069—1074)
|
| [4] |
Du T., Long Y., Tang Q., Chem. J. Chinese Universities,2017, 38(2),225—230
|
|
(杜涛, 龙渊, 汤琦,.高等学校化学学报, 2017, 38(2), 225—230)
|
| [5] |
Lu H. S., Bai L., Xiong W. W., Li P., Ding J., Zhang G., Wu T., Zhao Y., Lee J. M., Yang Y., Geng B., Zhang Q., Inorg. Chem., 2014, 53, 8529—8537
|
| [6] |
Gao J., Ye K., Yang L., Xiong W. W., Ye L., Wang Y., Zhang Q., Inorg. Chem., 2014, 53, 691—693
|
| [7] |
Zhang W., Lu G., Cui C., Liu Y., Li S., Yan W., Xing C., Chi Y. R., Yang Y., Huo F., Adv. Mater., 2014, 26, 4056—4060
|
| [8] |
Zhu Q. L., Li J., Xu Q., J. Am. Chem. Soc., 2013, 135, 10210—10213
|
| [9] |
Lu G., Li S., Guo Z., Farha O. K., Hauser B. G., Qi X., Wang Y., Wang X., Han S., Liu X., DuChene J. S., Zhang H., Zhang Q., Chen X., Ma J., Loo S. C., Wei W. D., Yang Y., Hupp J. T., Huo F., Nat. Chem., 2012, 4, 310—316
|
| [10] |
Sumida K., Rogow D. L., Mason J. A., McDonald T. M., Bloch E. D., Herm Z. R., Bae T. H., Long J. R., Chem. Rev., 2012, 112, 724—781
|
| [11] |
Schröder F., Esken D., Cokoja M., van den Berg M. W., Lebedev O. I., van Tendeloo G., Walaszek B., Buntkowsky G., Limbach H. H., Chaudret B., J. Am. Chem. Soc., 2008, 130, 6119—6130
|
| [12] |
Sabo M., Henschel A., Fröde H., Klemm E., Kaskel S., J. Mater. Chem., 2007, 17, 3827—3832
|
| [13] |
Proch S., Herrmannsdörfer J., Kempe R., Kern C., Jess A., Seyfarth L., Senker J., Chem. Eur. J., 2008, 14, 8204—8212
|
| [14] |
Park Y. K., Choi S. B., Nam H. J., Jung D. Y., Ahn H. C., Choi K., Furukawa H., Kim J., Chem. Commun., 2010, 46, 3086—3088
|
| [15] |
Huang Y., Zheng Z., Liu T., Lü J., Lin Z., Li H., Cao R., Catal. Commun., 2011, 14, 27—31
|
| [16] |
Hermes S., Schröter M. K., Schmid R., Khodeir L., Muhler M., Tissler A., Fischer R. W., Fischer R. A., Angew. Chem. Int. Ed., 2005, 44, 6237—6241
|
| [17] |
Esken D., Turner S., Lebedev O. I., van Tendeloo G., Fischer R. A., Chem. Mater., 2010, 22, 6393—6401
|
| [18] |
Dhakshinamoorthy A., Garcia H., Chem. Soc. Rev., 2012, 41, 5262—5284
|
| [19] |
Burtch N. C., Jasuja H., Walton K. S., Chem. Rev., 2014, 114, 10575—10612
|
| [20] |
Bo X. F., Wu P. Y., Liu D. H., Yang Q. Y., Ma Q. T., Lan L., Wang S. H., Zhang Y., Zhong C. L., J. Chem. Ind.Eng.(China), 2014, 65, 1644—1651
|
|
(薄晓帆, 吴平易, 刘大欢, 阳庆元, 麻沁甜, 兰玲, 王少华, 张轶, 仲崇立. 化工学报, 2014, 65, 1644—1651)
|
| [21] |
Vilhelmsen L. B., Walton K. S., Sholl D. S., J. Am. Chem. Soc., 2012, 134, 12807—12816
|
| [22] |
Vilhelmsen L. B., Sholl D. S., J. Phys. Chem. Lett., 2012, 3, 3702—3706
|
| [23] |
Cavka J. H., Jakobsen S., Olsbye U., Guillou N., Lamberti C., Bordiga S., Lillerud K. P., J. Am. Chem. Soc., 2008, 130, 13850—13851
|
| [24] |
Wu R., Qian X., Zhou K., Liu H., Yadian B., Wei J., Zhu H., Huang Y., J. Mater. Chem. A,2013, 1, 14294—14299
|
| [25] |
Dong W., Feng C., Zhang L., Shang N., Gao S., Wang C., Wang Z., Catal. Lett., 2016, 146, 117—125
|
| [26] |
Chen D. L., Wu S., Yang P., He S., Dou L., Wang F. F., J. Phys. Chem.C,2017, 121, 8857—8863
|
| [27] |
Larsen A. H., Kleis J., Thygesen K. S., Nørskov J. K., Jacobsen K. W., Phys. Rev.B,2011, 84, 245429
|
| [28] |
Wang L. L., Johnson D. D., J. Am. Chem. Soc., 2007, 129, 3658—3664
|
| [29] |
Wu H., Chua Y. S., Krungleviciute V., Madhusudan T., Chen P., Yildirim T., Zhou W., J. Am. Chem. Soc., 2013, 135(28), 10525—10532
|
| [30] |
Tang W., Sanville E., Henkelman G., J. Phys.: Condens.Matter,2009, 21, 084204
|
| [31] |
Sanville E., Kenny S. D., Smith R., Henkelman G., J. Comput. Chem., 2007, 28, 899—908
|
 )
)