

高等学校化学学报 ›› 2021, Vol. 42 ›› Issue (9): 2752.doi: 10.7503/cjcu20210180
黄罗仪1, 翁约约1,2, 黄旭慧3, 王朝杰1( )
)
收稿日期:2020-03-16
出版日期:2021-09-10
发布日期:2021-05-13
通讯作者:
王朝杰
E-mail:chjwang@wmu.edu.cn
基金资助:
HUANG Luoyi1, WENG Yueyue1,2, HUANG Xuhui3, WANG Chaojie1()
Received:2020-03-16
Online:2021-09-10
Published:2021-05-13
Contact:
WANG Chaojie
E-mail:chjwang@wmu.edu.cn
Supported by:摘要:
采用密度泛函理论中ωB97XD/6-311+G(2d,p)方法对车前草中的黄酮类化合物进行量化计算研究, 探讨了该类化合物的构效关系. 首先研究了13种车前草中提取的黄酮类化合物(1~13)和已成药的二氢杨梅素(14)的几何结构、 谱学性质及电子结构, 再运用概念密度泛函理论进行反应指数分析, 最后利用药代动力学平台开展了成药性评价. 根据几何结构的分析结果初步推测14种黄酮类化合物的抗氧化能力强弱顺序为10>12≈ 7>13>6>4>14≈9≈8>11>5>3≈2>1. 核磁共振拟合结果表明, 利用该方法得到的核磁位移理论值与实验值吻合度较高(R2>0.95). 分子表面静电势图显示, 14种黄酮类化合物的静电势的极小值点都位于羰基氧附近, 极大值点都位于羟基氢附近, 且B环上对位羟基的极大值>C环和A环羟基极大值. 全局反应性描述符结果显示, 化合物1, 2, 4, 11和12的化学势较低, 电负性、 亲电指数和硬度较高, 说明这5种化合物的稳定性和反应性较好. 通过局部反应性描述符进一步预测了14种黄酮类化合物的亲核/亲电反应位点. 药代动力学结果显示, 化合物1, 3, 4, 6, 12和13的成药性和药代动力学活性较好. 研究结果表明, 化合物4和12最有成药潜力, 可进行更深入的实验研究.
中图分类号:
TrendMD:
黄罗仪, 翁约约, 黄旭慧, 王朝杰. 车前草中黄酮类成分结构和性质的理论研究. 高等学校化学学报, 2021, 42(9): 2752.
HUANG Luoyi, WENG Yueyue, HUANG Xuhui, WANG Chaojie. Theoretical Study on the Structures and Properties of Flavonoids in Plantain. Chem. J. Chinese Universities, 2021, 42(9): 2752.
Fig.1 2D?structural formulas of 14 flavonoids
| Compd. | Bond | Length/nm | Compd. | Bond | Length/nm | Compd. | Bond | Length/nm |
|---|---|---|---|---|---|---|---|---|
| 1 | O19—H29 | 0.170 | 7 | O17—H30 | 0.177 | 11 | O19—H30 | 0.169 |
| 2 | O19—H29 | 0.170 | O17—H35 | 0.200 | O21—H29 | 0.208 | ||
| O21—H30 | 0.215 | O21—H31 | 0.210 | 12 | O19—H30 | 0.167 | ||
| 3 | O19—H31 | 0.171 | 8 | O18—H30 | 0.225 | O20—H35 | 0.215 | |
| O20—H33 | 0.215 | O19—H29 | 0.170 | O21—H29 | 0.208 | |||
| 4 | O17—H31 | 0.171 | O20—H28 | 0.216 | 13 | O17—H29 | 0.170 | |
| O20—H34 | 0.218 | 9 | O18—H31 | 0.225 | O19—H30 | 0.208 | ||
| O21—H32 | 0.220 | O19—H29 | 0.170 | O22—H31 | 0.215 | |||
| 5 | O17—H29 | 0.177 | O21—H28 | 0.216 | 14 | O17—H31 | 0.173 | |
| O17—H31 | 0.201 | 10 | O18—H30 | 0.225 | O20—H34 | 0.218 | ||
| 6 | O19—H29 | 0.177 | O19—H29 | 0.170 | O21—H32 | 0.220 | ||
| O19—H31 | 0.201 | O20—H18 | 0.216 | |||||
| O20—H32 | 0.215 | O22—H31 | 0.215 |
Table 1 Hydrogen bond parameters of 14 flavonoids
| Compd. | Bond | Length/nm | Compd. | Bond | Length/nm | Compd. | Bond | Length/nm |
|---|---|---|---|---|---|---|---|---|
| 1 | O19—H29 | 0.170 | 7 | O17—H30 | 0.177 | 11 | O19—H30 | 0.169 |
| 2 | O19—H29 | 0.170 | O17—H35 | 0.200 | O21—H29 | 0.208 | ||
| O21—H30 | 0.215 | O21—H31 | 0.210 | 12 | O19—H30 | 0.167 | ||
| 3 | O19—H31 | 0.171 | 8 | O18—H30 | 0.225 | O20—H35 | 0.215 | |
| O20—H33 | 0.215 | O19—H29 | 0.170 | O21—H29 | 0.208 | |||
| 4 | O17—H31 | 0.171 | O20—H28 | 0.216 | 13 | O17—H29 | 0.170 | |
| O20—H34 | 0.218 | 9 | O18—H31 | 0.225 | O19—H30 | 0.208 | ||
| O21—H32 | 0.220 | O19—H29 | 0.170 | O22—H31 | 0.215 | |||
| 5 | O17—H29 | 0.177 | O21—H28 | 0.216 | 14 | O17—H31 | 0.173 | |
| O17—H31 | 0.201 | 10 | O18—H30 | 0.225 | O20—H34 | 0.218 | ||
| 6 | O19—H29 | 0.177 | O19—H29 | 0.170 | O21—H32 | 0.220 | ||
| O19—H31 | 0.201 | O20—H18 | 0.216 | |||||
| O20—H32 | 0.215 | O22—H31 | 0.215 |
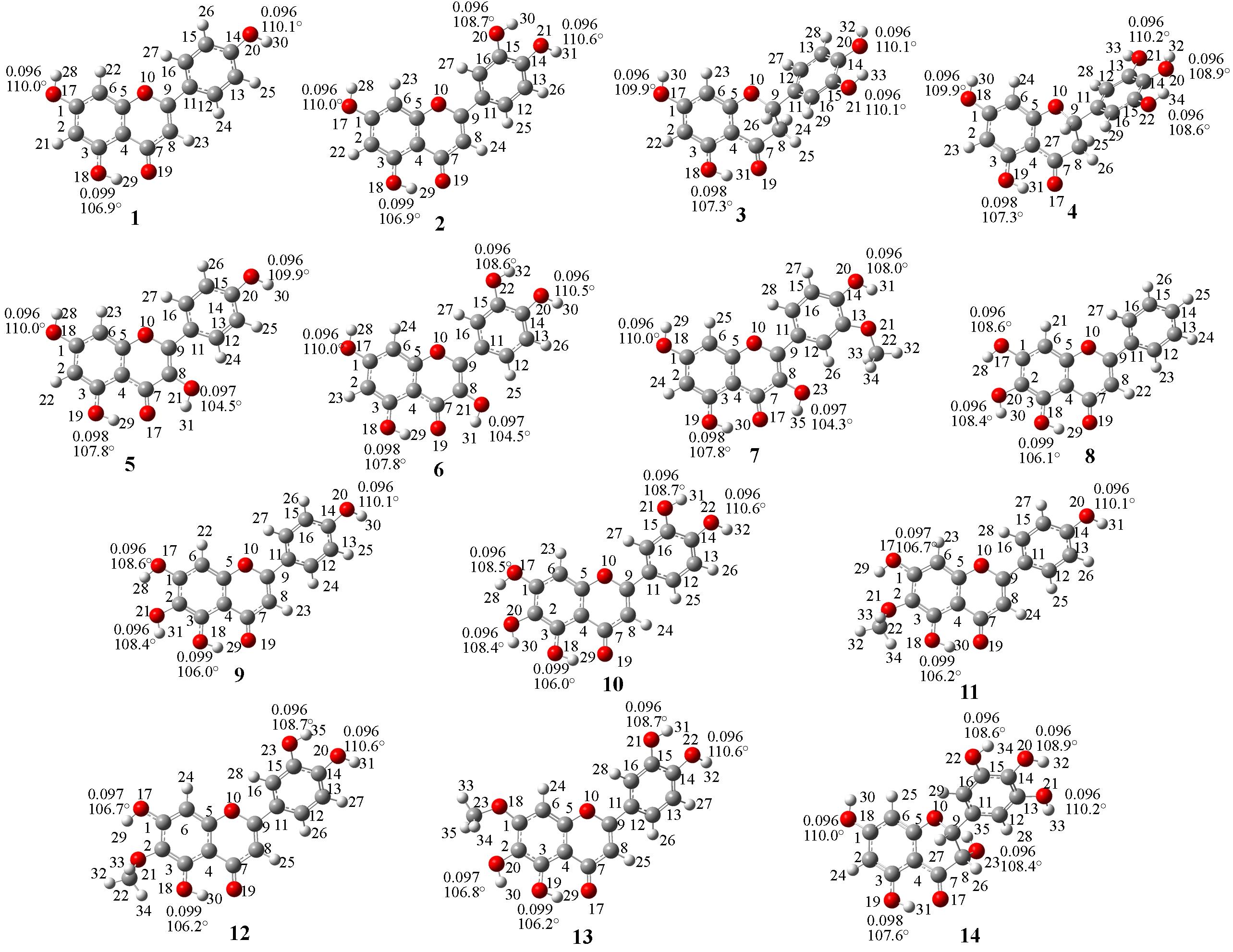
Fig.2 Optimized geometric structures of 14 flavonoids at the level of ωB970XD/6?311+G(2d, p) in vacuum
| 1 | Theo. | Exp.[ | 2 | Theo. | Exp.[ | 4 | Theo. | Exp.[ | 5 | Theo. | Exp.[ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 170.3 | 166.2 | C1 | 170.3 | 161.9 | C1 | 172.8 | 166.8 | C1 | 170.8 | 165.7 |
| C2 | 101.5 | 100.4 | C2 | 101.4 | 99.3 | C2 | 97.9 | 163.4 | C2 | 100.7 | 99.4 |
| C3 | 170.2 | 163.2 | C3 | 170.3 | 164.4 | C3 | 172.4 | 163.4 | C3 | 169.2 | 162.6 |
| C4 | 108.4 | 105.0 | C4 | 108.5 | 103.3 | C4 | 105.6 | 102.0 | C4 | 106.4 | 104.6 |
| C5 | 164.2 | 159.5 | C5 | 164.3 | 157.7 | C5 | 171.3 | 163.0 | C5 | 163.2 | 158.4 |
| C6 | 96.1 | 95.3 | C6 | 96.1 | 94.3 | C6 | 97.0 | 95.1 | C6 | 96.1 | 94.6 |
| C7 | 190.4 | 183.8 | C7 | 190.3 | 182.1 | C7 | 204.9 | 196.4 | C7 | 182.2 | 177.5 |
| C8 | 108.2 | 103.7 | C8 | 108.7 | 104.1 | C8 | 49.3 | 40.4 | C8 | 140.4 | 137.3 |
| C9 | 172.6 | 166.3 | C9 | 172.5 | 164.6 | C9 | 82.3 | 78.6 | C9 | 150.6 | 148.2 |
| C11 | 128.2 | 123.3 | C11 | 130.2 | 121.9 | C11 | 139.3 | 128.7 | C11 | 127.7 | 123.9 |
| C12 | 136.1 | 129.4 | C12 | 125.6 | 113.8 | C12 | 105.9 | 109.3 | C12 | 138.1 | 130.8 |
| C13 | 119.5 | 117.1 | C13 | 119.5 | 116.5 | C13 | 149.4 | 145.7 | C13 | 118.5 | 116.5 |
| C14 | 167.1 | 162.9 | C14 | 152.4 | 146.2 | C14 | 135.8 | 135.3 | C14 | 165.4 | 160.5 |
| C15 | 120.2 | 117.1 | C15 | 151.1 | 150.2 | C15 | 150.5 | 145.8 | C15 | 120.0 | 116.5 |
| C16 | 136.0 | 129.4 | C16 | 118.8 | 119.4 | C16 | 109.5 | 106.5 | C16 | 134.9 | 130.8 |
| H21 | 6.6 | 6.2 | H22 | 6.6 | 6.2 | H23 | 6.2 | 6.0 | H22 | 6.6 | 6.4 |
| H22 | 6.6 | 6.4 | H23 | 6.7 | 6.5 | H24 | 6.1 | 6.0 | H23 | 6.7 | 6.2 |
| H23 | 6.8 | 6.6 | H24 | 6.8 | 6.7 | H25 | 2.8 | 2.8 | H24 | 9.1 | 8.1 |
| H24 | 8.2 | 7.8 | H25 | 7.7 | 7.4 | H26 | 2.5 | H25 | 7.3 | 6.9 | |
| H25 | 7.2 | 6.9 | H26 | 7.2 | 6.9 | H27 | 5.3 | 5.4 | H26 | 7.5 | 6.9 |
| H26 | 7.4 | 6.9 | H27 | 8.0 | 7.4 | H28 | 7.0 | 6.8 | H27 | 8.6 | 8.1 |
| H27 | 8.5 | 7.8 | H28 | 5.1 | H29 | 6.7 | 6.9 | H28 | 5.0 | ||
| H28 | 5.1 | H29 | 12.8 | 13.0 | H30 | 4.9 | H29 | 11.5 | |||
| H29 | 12.8 | H30 | 5.3 | H31 | 12.0 | H30 | 4.8 | ||||
| H30 | 5.0 | H31 | 5.0 | H32 | 5.0 | H31 | 6.6 | ||||
| H33 | 4.6 | ||||||||||
| 6 | Theo. | Exp.[ | 7 | Theo. | Exp.[ | 8 | Theo. | Exp.[ | 9 | Theo. | Exp.[ |
| C1 | 170.8 | 164.1 | C1 | 170.8 | 163.9 | C1 | 157.2 | 153.6 | C1 | 156.9 | 153.8 |
| C2 | 100.6 | 98.6 | C2 | 100.6 | 98.2 | C2 | 132.3 | 129.4 | C2 | 132.2 | 129.6 |
| C3 | 169.1 | 161.2 | C3 | 169.5 | 160.6 | C3 | 151.5 | 146.6 | C3 | 151.3 | 147.5 |
| C4 | 106.3 | 104.0 | C4 | 106.1 | 103.0 | C4 | 107.9 | 104.5 | C4 | 107.7 | 104.5 |
| C5 | 163.3 | 157.2 | C5 | 163.1 | 156.1 | C5 | 157.0 | 150.6 | C5 | 156.7 | 150.1 |
| C6 | 95.9 | 93.5 | C6 | 95.9 | 93.5 | C6 | 97.2 | 93.7 | C6 | 97.1 | 94.3 |
| C7 | 182.2 | 177.7 | C7 | 182.3 | 175.8 | C7 | 189.8 | 182.7 | C7 | 189.8 | 182.5 |
| C8 | 141.1 | 134.2 | C8 | 140.5 | 135.8 | C8 | 109.8 | 104.0 | C8 | 107.8 | 102.7 |
| C9 | 150.1 | 156.4 | C9 | 150.4 | 146.6 | C9 | 173.3 | 164.1 | C9 | 173.0 | 164.0 |
| C11 | 129.8 | 120.7 | C11 | 127.6 | 121.7 | C11 | 138.1 | 131.4 | C11 | 128.9 | 121.9 |
| C12 | 127.8 | 120.9 | C12 | 115.9 | 111.8 | C12 | 133.5 | 125.8 | C12 | 136.0 | 128.8 |
| C13 | 118.8 | 115.6 | C13 | 151.4 | 147.3 | C13 | 134.8 | 129.0 | C13 | 119.1 | 116.4 |
| C14 | 150.7 | 148.3 | C14 | 154.6 | 148.8 | C14 | 138.4 | 131.6 | C14 | 166.8 | 161.5 |
| C15 | 150.9 | 145.1 | C15 | 118.7 | 115.5 | C15 | 134.8 | 129.0 | C15 | 120.0 | 116.4 |
| C16 | 117.7 | 115.4 | C16 | 125.1 | 121.9 | C16 | 133.0 | 125.8 | C16 | 135.7 | 128.8 |
| H23 | 6.6 | 6.2 | C22 | 56.1 | 55.8 | H21 | 7.0 | 6.6 | H22 | 6.9 | 6.6 |
| H24 | 6.7 | 6.4 | H24 | 6.6 | 6.2 | H22 | 6.8 | 6.8 | H23 | 6.7 | 6.8 |
| H25 | 8.6 | 7.3 | H25 | 6.7 | 6.5 | H23 | 8.3 | 8.0 | H24 | 8.2 | 7.9 |
| H26 | 7.3 | 6.9 | H26 | 8.8 | 7.7 | H24 | 8.0 | 7.7 | H25 | 7.2 | 6.9 |
| H27 | 8.1 | 7.3 | H27 | 7.5 | 6.9 | H25 | 8.1 | 7.7 | H26 | 7.4 | 6.9 |
| H28 | 5.1 | 12.7 | H28 | 8.2 | 7.8 | H26 | 8.0 | 7.7 | H27 | 8.4 | 7.9 |
| H29 | 11.5 | 10.8 | H29 | 5.1 | 10.7 | H27 | 8.5 | 8.0 | H28 | 5.7 | 10.5 |
| H30 | 4.8 | 9.7 | H30 | 11.6 | 12.5 | H28 | 5.8 | 10.6 | H29 | 12.7 | 12.8 |
| 6 | Theo. | Exp.[ | 7 | Theo. | Exp.[ | 8 | Theo. | Exp.[ | 9 | Theo. | Exp.[ |
| H31 | 6.7 | H31 | 5.8 | 9.7 | H29 | 12.7 | 12.7 | H30 | 4.9 | 10.3 | |
| H32 | 5.3 | 9.3 | H32 | 4.2 | 3.9 | H30 | 5.0 | 10.0 | H31 | 5.0 | 9.2 |
| H33 | 3.9 | 3.9 | |||||||||
| H34 | 3.9 | 3.9 | |||||||||
| H35 | 6.8 | 9.4 | |||||||||
| 10 | Theo. | Exp.[ | 11 | Theo. | Exp.[ | 12 | Theo. | Exp.[ | 13 | Theo. | Exp.[ |
| C1 | 156.8 | 154.3 | C1 | 163.0 | 157.3 | C1 | 162.9 | 156.5 | C1 | 159.4 | 161.0 |
| C2 | 132.0 | 129.9 | C2 | 133.7 | 131.4 | C2 | 133.6 | 132.5 | C2 | 140.4 | 158.0 |
| C3 | 151.3 | 149.6 | C3 | 159.7 | 152.8 | C3 | 159.8 | 152.1 | C3 | 152.6 | 160.8 |
| C4 | 107.8 | 105.0 | C4 | 108.1 | 104.1 | C4 | 108.2 | 105.7 | C4 | 111.0 | 105.5 |
| C5 | 156.8 | 146.2 | C5 | 159.9 | 152.4 | C5 | 159.9 | 152.4 | C5 | 155.0 | 162.7 |
| C6 | 97.1 | 91.0 | C6 | 96.0 | 94.3 | C6 | 96.1 | 94.3 | C6 | 106.1 | 96.5 |
| C7 | 189.7 | 182.0 | C7 | 190.1 | 182.1 | C7 | 190.0 | 182.2 | C7 | 191.0 | 181.8 |
| C8 | 108.5 | 102.5 | C8 | 107.9 | 102.4 | C8 | 108.7 | 102.7 | C8 | 107.9 | 102.4 |
| C9 | 173.0 | 163.3 | C9 | 172.5 | 163.8 | C9 | 172.5 | 164.3 | C9 | 173.5 | 164.4 |
| C11 | 131.2 | 121.6 | C11 | 128.6 | 121.2 | C11 | 131.0 | 121.1 | C11 | 130.3 | 121.1 |
| C12 | 125.3 | 118.8 | C12 | 136.0 | 128.5 | C12 | 125.3 | 128.0 | C12 | 125.7 | 118.4 |
| C13 | 119.4 | 116.0 | C13 | 119.1 | 116.0 | C13 | 119.3 | 116.0 | C13 | 119.3 | 115.7 |
| C14 | 152.0 | 149.7 | C14 | 166.8 | 161.2 | C14 | 151.9 | 161.4 | C14 | 152.3 | 149.7 |
| C15 | 150.9 | 145.8 | C15 | 120.0 | 116.0 | C15 | 150.9 | 116.0 | C15 | 150.9 | 145.9 |
| C16 | 118.7 | 113.4 | C16 | 135.7 | 128.5 | C16 | 118.7 | 128.6 | C16 | 119.1 | 114.1 |
| H23 | 6.9 | 6.7 | C22 | 60.6 | 60.0 | C22 | 60.5 | 60.3 | C23 | 62.0 | |
| H24 | 6.7 | 6.9 | H23 | 6.9 | 6.6 | H24 | 6.9 | 7.0 | H24 | 7.1 | 6.8 |
| H25 | 7.7 | 7.5 | H24 | 6.7 | 6.8 | H25 | 6.7 | 6.9 | H25 | 6.8 | 6.7 |
| H26 | 7.3 | 6.9 | H25 | 8.2 | 7.9 | H26 | 7.7 | 7.9 | H26 | 7.7 | 7.4 |
| H27 | 7.9 | 7.4 | H26 | 7.2 | 6.9 | H27 | 7.3 | 6.9 | H27 | 7.3 | 6.9 |
| H28 | 5.8 | H27 | 7.4 | 6.9 | H28 | 7.9 | 7.9 | H28 | 8.0 | 7.5 | |
| H29 | 12.7 | H28 | 8.4 | 7.9 | H29 | 6.5 | H29 | 12.7 | 13.0 | ||
| H30 | 5.0 | H29 | 6.5 | H30 | 13.1 | 13.0 | H30 | 5.1 | |||
| H31 | 5.3 | H30 | 13.1 | 13.1 | H31 | 4.9 | 10.4 | H31 | 5.4 | 9.4 | |
| H32 | 4.9 | H31 | 4.9 | H32 | 4.0 | 3.8 | H32 | 5.0 | 9.9 | ||
| H32 | 4.0 | 3.8 | H33 | 3.5 | 3.8 | H33 | 4.0 | 3.9 | |||
| H33 | 3.5 | 3.8 | H34 | 4.9 | 3.8 | H34 | 4.6 | 3.9 | |||
| H34 | 4.9 | 3.8 | H35 | 5.3 | 6.9 | H35 | 3.6 | 3.9 |
Table 2 Theoretical and experimental values of 13C NMR and 1H NMR of the compound at the level of ωB97XD//GIAO/6-311+G(2d, p) in DMSO
| 1 | Theo. | Exp.[ | 2 | Theo. | Exp.[ | 4 | Theo. | Exp.[ | 5 | Theo. | Exp.[ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 170.3 | 166.2 | C1 | 170.3 | 161.9 | C1 | 172.8 | 166.8 | C1 | 170.8 | 165.7 |
| C2 | 101.5 | 100.4 | C2 | 101.4 | 99.3 | C2 | 97.9 | 163.4 | C2 | 100.7 | 99.4 |
| C3 | 170.2 | 163.2 | C3 | 170.3 | 164.4 | C3 | 172.4 | 163.4 | C3 | 169.2 | 162.6 |
| C4 | 108.4 | 105.0 | C4 | 108.5 | 103.3 | C4 | 105.6 | 102.0 | C4 | 106.4 | 104.6 |
| C5 | 164.2 | 159.5 | C5 | 164.3 | 157.7 | C5 | 171.3 | 163.0 | C5 | 163.2 | 158.4 |
| C6 | 96.1 | 95.3 | C6 | 96.1 | 94.3 | C6 | 97.0 | 95.1 | C6 | 96.1 | 94.6 |
| C7 | 190.4 | 183.8 | C7 | 190.3 | 182.1 | C7 | 204.9 | 196.4 | C7 | 182.2 | 177.5 |
| C8 | 108.2 | 103.7 | C8 | 108.7 | 104.1 | C8 | 49.3 | 40.4 | C8 | 140.4 | 137.3 |
| C9 | 172.6 | 166.3 | C9 | 172.5 | 164.6 | C9 | 82.3 | 78.6 | C9 | 150.6 | 148.2 |
| C11 | 128.2 | 123.3 | C11 | 130.2 | 121.9 | C11 | 139.3 | 128.7 | C11 | 127.7 | 123.9 |
| C12 | 136.1 | 129.4 | C12 | 125.6 | 113.8 | C12 | 105.9 | 109.3 | C12 | 138.1 | 130.8 |
| C13 | 119.5 | 117.1 | C13 | 119.5 | 116.5 | C13 | 149.4 | 145.7 | C13 | 118.5 | 116.5 |
| C14 | 167.1 | 162.9 | C14 | 152.4 | 146.2 | C14 | 135.8 | 135.3 | C14 | 165.4 | 160.5 |
| C15 | 120.2 | 117.1 | C15 | 151.1 | 150.2 | C15 | 150.5 | 145.8 | C15 | 120.0 | 116.5 |
| C16 | 136.0 | 129.4 | C16 | 118.8 | 119.4 | C16 | 109.5 | 106.5 | C16 | 134.9 | 130.8 |
| H21 | 6.6 | 6.2 | H22 | 6.6 | 6.2 | H23 | 6.2 | 6.0 | H22 | 6.6 | 6.4 |
| H22 | 6.6 | 6.4 | H23 | 6.7 | 6.5 | H24 | 6.1 | 6.0 | H23 | 6.7 | 6.2 |
| H23 | 6.8 | 6.6 | H24 | 6.8 | 6.7 | H25 | 2.8 | 2.8 | H24 | 9.1 | 8.1 |
| H24 | 8.2 | 7.8 | H25 | 7.7 | 7.4 | H26 | 2.5 | H25 | 7.3 | 6.9 | |
| H25 | 7.2 | 6.9 | H26 | 7.2 | 6.9 | H27 | 5.3 | 5.4 | H26 | 7.5 | 6.9 |
| H26 | 7.4 | 6.9 | H27 | 8.0 | 7.4 | H28 | 7.0 | 6.8 | H27 | 8.6 | 8.1 |
| H27 | 8.5 | 7.8 | H28 | 5.1 | H29 | 6.7 | 6.9 | H28 | 5.0 | ||
| H28 | 5.1 | H29 | 12.8 | 13.0 | H30 | 4.9 | H29 | 11.5 | |||
| H29 | 12.8 | H30 | 5.3 | H31 | 12.0 | H30 | 4.8 | ||||
| H30 | 5.0 | H31 | 5.0 | H32 | 5.0 | H31 | 6.6 | ||||
| H33 | 4.6 | ||||||||||
| 6 | Theo. | Exp.[ | 7 | Theo. | Exp.[ | 8 | Theo. | Exp.[ | 9 | Theo. | Exp.[ |
| C1 | 170.8 | 164.1 | C1 | 170.8 | 163.9 | C1 | 157.2 | 153.6 | C1 | 156.9 | 153.8 |
| C2 | 100.6 | 98.6 | C2 | 100.6 | 98.2 | C2 | 132.3 | 129.4 | C2 | 132.2 | 129.6 |
| C3 | 169.1 | 161.2 | C3 | 169.5 | 160.6 | C3 | 151.5 | 146.6 | C3 | 151.3 | 147.5 |
| C4 | 106.3 | 104.0 | C4 | 106.1 | 103.0 | C4 | 107.9 | 104.5 | C4 | 107.7 | 104.5 |
| C5 | 163.3 | 157.2 | C5 | 163.1 | 156.1 | C5 | 157.0 | 150.6 | C5 | 156.7 | 150.1 |
| C6 | 95.9 | 93.5 | C6 | 95.9 | 93.5 | C6 | 97.2 | 93.7 | C6 | 97.1 | 94.3 |
| C7 | 182.2 | 177.7 | C7 | 182.3 | 175.8 | C7 | 189.8 | 182.7 | C7 | 189.8 | 182.5 |
| C8 | 141.1 | 134.2 | C8 | 140.5 | 135.8 | C8 | 109.8 | 104.0 | C8 | 107.8 | 102.7 |
| C9 | 150.1 | 156.4 | C9 | 150.4 | 146.6 | C9 | 173.3 | 164.1 | C9 | 173.0 | 164.0 |
| C11 | 129.8 | 120.7 | C11 | 127.6 | 121.7 | C11 | 138.1 | 131.4 | C11 | 128.9 | 121.9 |
| C12 | 127.8 | 120.9 | C12 | 115.9 | 111.8 | C12 | 133.5 | 125.8 | C12 | 136.0 | 128.8 |
| C13 | 118.8 | 115.6 | C13 | 151.4 | 147.3 | C13 | 134.8 | 129.0 | C13 | 119.1 | 116.4 |
| C14 | 150.7 | 148.3 | C14 | 154.6 | 148.8 | C14 | 138.4 | 131.6 | C14 | 166.8 | 161.5 |
| C15 | 150.9 | 145.1 | C15 | 118.7 | 115.5 | C15 | 134.8 | 129.0 | C15 | 120.0 | 116.4 |
| C16 | 117.7 | 115.4 | C16 | 125.1 | 121.9 | C16 | 133.0 | 125.8 | C16 | 135.7 | 128.8 |
| H23 | 6.6 | 6.2 | C22 | 56.1 | 55.8 | H21 | 7.0 | 6.6 | H22 | 6.9 | 6.6 |
| H24 | 6.7 | 6.4 | H24 | 6.6 | 6.2 | H22 | 6.8 | 6.8 | H23 | 6.7 | 6.8 |
| H25 | 8.6 | 7.3 | H25 | 6.7 | 6.5 | H23 | 8.3 | 8.0 | H24 | 8.2 | 7.9 |
| H26 | 7.3 | 6.9 | H26 | 8.8 | 7.7 | H24 | 8.0 | 7.7 | H25 | 7.2 | 6.9 |
| H27 | 8.1 | 7.3 | H27 | 7.5 | 6.9 | H25 | 8.1 | 7.7 | H26 | 7.4 | 6.9 |
| H28 | 5.1 | 12.7 | H28 | 8.2 | 7.8 | H26 | 8.0 | 7.7 | H27 | 8.4 | 7.9 |
| H29 | 11.5 | 10.8 | H29 | 5.1 | 10.7 | H27 | 8.5 | 8.0 | H28 | 5.7 | 10.5 |
| H30 | 4.8 | 9.7 | H30 | 11.6 | 12.5 | H28 | 5.8 | 10.6 | H29 | 12.7 | 12.8 |
| 6 | Theo. | Exp.[ | 7 | Theo. | Exp.[ | 8 | Theo. | Exp.[ | 9 | Theo. | Exp.[ |
| H31 | 6.7 | H31 | 5.8 | 9.7 | H29 | 12.7 | 12.7 | H30 | 4.9 | 10.3 | |
| H32 | 5.3 | 9.3 | H32 | 4.2 | 3.9 | H30 | 5.0 | 10.0 | H31 | 5.0 | 9.2 |
| H33 | 3.9 | 3.9 | |||||||||
| H34 | 3.9 | 3.9 | |||||||||
| H35 | 6.8 | 9.4 | |||||||||
| 10 | Theo. | Exp.[ | 11 | Theo. | Exp.[ | 12 | Theo. | Exp.[ | 13 | Theo. | Exp.[ |
| C1 | 156.8 | 154.3 | C1 | 163.0 | 157.3 | C1 | 162.9 | 156.5 | C1 | 159.4 | 161.0 |
| C2 | 132.0 | 129.9 | C2 | 133.7 | 131.4 | C2 | 133.6 | 132.5 | C2 | 140.4 | 158.0 |
| C3 | 151.3 | 149.6 | C3 | 159.7 | 152.8 | C3 | 159.8 | 152.1 | C3 | 152.6 | 160.8 |
| C4 | 107.8 | 105.0 | C4 | 108.1 | 104.1 | C4 | 108.2 | 105.7 | C4 | 111.0 | 105.5 |
| C5 | 156.8 | 146.2 | C5 | 159.9 | 152.4 | C5 | 159.9 | 152.4 | C5 | 155.0 | 162.7 |
| C6 | 97.1 | 91.0 | C6 | 96.0 | 94.3 | C6 | 96.1 | 94.3 | C6 | 106.1 | 96.5 |
| C7 | 189.7 | 182.0 | C7 | 190.1 | 182.1 | C7 | 190.0 | 182.2 | C7 | 191.0 | 181.8 |
| C8 | 108.5 | 102.5 | C8 | 107.9 | 102.4 | C8 | 108.7 | 102.7 | C8 | 107.9 | 102.4 |
| C9 | 173.0 | 163.3 | C9 | 172.5 | 163.8 | C9 | 172.5 | 164.3 | C9 | 173.5 | 164.4 |
| C11 | 131.2 | 121.6 | C11 | 128.6 | 121.2 | C11 | 131.0 | 121.1 | C11 | 130.3 | 121.1 |
| C12 | 125.3 | 118.8 | C12 | 136.0 | 128.5 | C12 | 125.3 | 128.0 | C12 | 125.7 | 118.4 |
| C13 | 119.4 | 116.0 | C13 | 119.1 | 116.0 | C13 | 119.3 | 116.0 | C13 | 119.3 | 115.7 |
| C14 | 152.0 | 149.7 | C14 | 166.8 | 161.2 | C14 | 151.9 | 161.4 | C14 | 152.3 | 149.7 |
| C15 | 150.9 | 145.8 | C15 | 120.0 | 116.0 | C15 | 150.9 | 116.0 | C15 | 150.9 | 145.9 |
| C16 | 118.7 | 113.4 | C16 | 135.7 | 128.5 | C16 | 118.7 | 128.6 | C16 | 119.1 | 114.1 |
| H23 | 6.9 | 6.7 | C22 | 60.6 | 60.0 | C22 | 60.5 | 60.3 | C23 | 62.0 | |
| H24 | 6.7 | 6.9 | H23 | 6.9 | 6.6 | H24 | 6.9 | 7.0 | H24 | 7.1 | 6.8 |
| H25 | 7.7 | 7.5 | H24 | 6.7 | 6.8 | H25 | 6.7 | 6.9 | H25 | 6.8 | 6.7 |
| H26 | 7.3 | 6.9 | H25 | 8.2 | 7.9 | H26 | 7.7 | 7.9 | H26 | 7.7 | 7.4 |
| H27 | 7.9 | 7.4 | H26 | 7.2 | 6.9 | H27 | 7.3 | 6.9 | H27 | 7.3 | 6.9 |
| H28 | 5.8 | H27 | 7.4 | 6.9 | H28 | 7.9 | 7.9 | H28 | 8.0 | 7.5 | |
| H29 | 12.7 | H28 | 8.4 | 7.9 | H29 | 6.5 | H29 | 12.7 | 13.0 | ||
| H30 | 5.0 | H29 | 6.5 | H30 | 13.1 | 13.0 | H30 | 5.1 | |||
| H31 | 5.3 | H30 | 13.1 | 13.1 | H31 | 4.9 | 10.4 | H31 | 5.4 | 9.4 | |
| H32 | 4.9 | H31 | 4.9 | H32 | 4.0 | 3.8 | H32 | 5.0 | 9.9 | ||
| H32 | 4.0 | 3.8 | H33 | 3.5 | 3.8 | H33 | 4.0 | 3.9 | |||
| H33 | 3.5 | 3.8 | H34 | 4.9 | 3.8 | H34 | 4.6 | 3.9 | |||
| H34 | 4.9 | 3.8 | H35 | 5.3 | 6.9 | H35 | 3.6 | 3.9 |
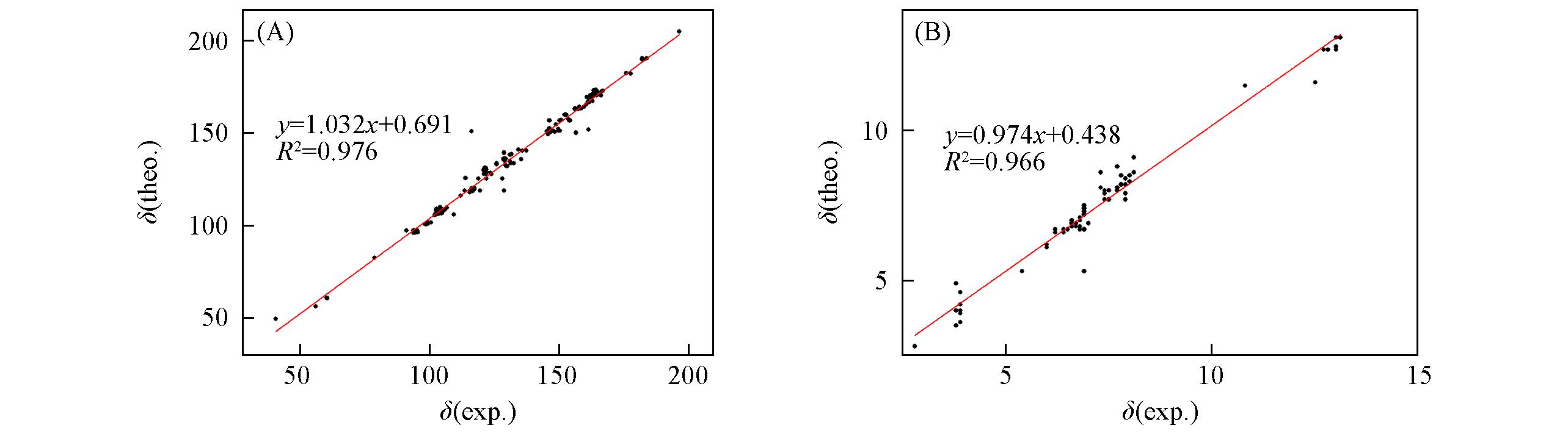
Fig.3 Correlation graphs between experimental and calculated 13C NMR(A) and 1H NMR(B) chemical shifts by ωB97XD/6?311+G(2d, p) method in DMSO
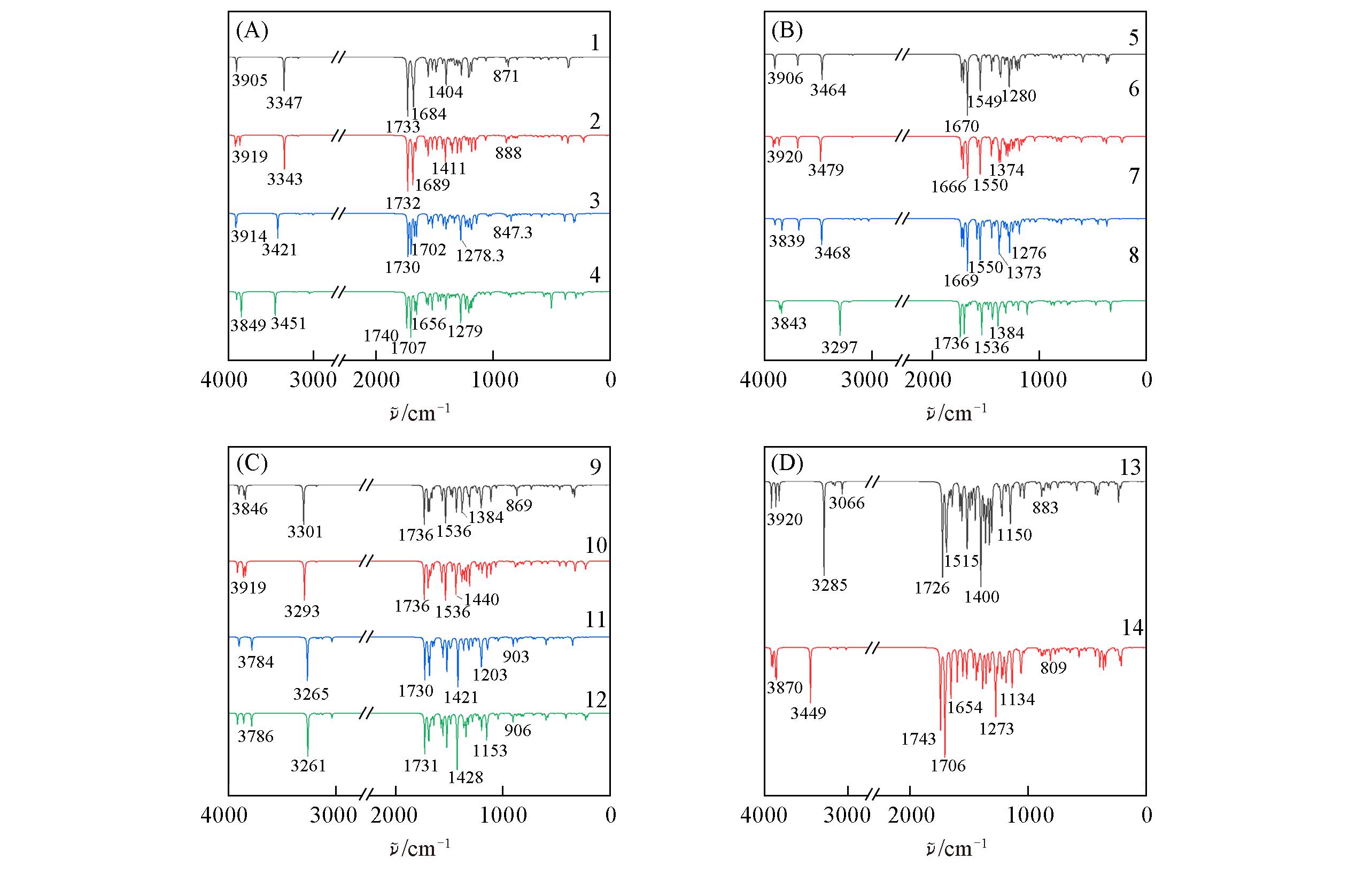
Fig.4 Calculated infrared spectra of 14 flavonoids by ωB97XD/6?311+G(2d, p) method in vacuum
| Compd. | E/eV | λ/nm | f | Configuration(%) | Compd. | E/eV | λ/nm | f | Configuration(%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6.3986 | 193.77 | 0.2331 | H-4→L+1 | 28.4 | 8 | 6.1950 | 200.14 | 0.3798 | H-1→L+3 | 19.0 |
| 5.3713 | 230.83 | 0.5704 | H-2→L | 30.3 | 5.1130 | 242.49 | 0.3887 | H→L+1 | 47.7 | ||
| 4.3488 | 285.10 | 0.4797 | H→L | 81.0 | 4.3673 | 283.89 | 0.4496 | H-1→L | 67.0 | ||
| 2 | 6.3847 | 194.19 | 0.3371 | H-5→L | 23.3 | 9 | 6.1917 | 200.24 | 0.5627 | H-2→L+1 | 30.9 |
| 5.3652 | 231.09 | 0.5613 | H-2→L | 30.2 | 5.1647 | 240.06 | 0.3556 | H→L+1 | 39.8 | ||
| 4.3338 | 286.08 | 0.4922 | H→L | 63.0 | 4.2466 | 291.96 | 0.6038 | H-1→L | 71.1 | ||
| 3 | 6.4021 | 193.66 | 0.4543 | H-3→L+1 | 55.8 | 10 | 6.1585 | 201.32 | 0.5873 | H-1→L+4 | 28.7 |
| 4.8064 | 257.96 | 0.3351 | H-2→L | 89.0 | 4.2134 | 294.26 | 0.4681 | H→L | 76.7 | ||
| 4 | 6.3085 | 196.54 | 0.5600 | H-1→L+4 | 31.9 | 11 | 6.3575 | 195.02 | 0.3613 | H-5→L | 22.3 |
| 4.8254 | 256.94 | 0.2768 | H-3→L | 83.7 | 5.3316 | 232.54 | 0.5412 | H→L+1 | 25.9 | ||
| 5 | 6.6481 | 186.50 | 0.4874 | H-3→L+2 | 27.7 | 4.3172 | 287.18 | 0.3724 | H→L | 81.2 | |
| 5.2108 | 237.94 | 0.5759 | H→L+1 | 23.0 | 12 | 6.3482 | 195.31 | 0.3164 | H-5→L | 34.4 | |
| 3.7785 | 328.13 | 0.6297 | H→L | 92.0 | 5.3256 | 232.81 | 0.5315 | H-1→L+1 | 36.0 | ||
| 6 | 6.0724 | 204.18 | 0.3920 | H-2→L+2 | 22.9 | 4.3063 | 287.91 | 0.3919 | H→L | 51.0 | |
| 5.2094 | 238.00 | 0.5679 | H→L+1 | 21.8 | 13 | 6.0731 | 204.15 | 0.5142 | H-1→L+4 | 26.4 | |
| 3.7664 | 329.18 | 0.6245 | H→L | 88.2 | 4.1614 | 297.94 | 0.5974 | H→L | 48.2 | ||
| 7 | 6.6384 | 186.77 | 0.3346 | H-3→L+3 | 24.5 | 14 | 6.2805 | 197.41 | 0.5505 | H-3→L+1 | 19.6 |
| 5.4868 | 225.97 | 0.4272 | H→L+2 | 29.4 | 4.7302 | 262.11 | 0.3216 | H-2→L | 53.8 | ||
| 3.7509 | 330.55 | 0.6480 | H→L | 86.7 | |||||||
Table 3 UV-Vis absorption spectral data of the 14 flavonoids in methanol by ωB97XD/6-311+G(2d, p) method*
| Compd. | E/eV | λ/nm | f | Configuration(%) | Compd. | E/eV | λ/nm | f | Configuration(%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6.3986 | 193.77 | 0.2331 | H-4→L+1 | 28.4 | 8 | 6.1950 | 200.14 | 0.3798 | H-1→L+3 | 19.0 |
| 5.3713 | 230.83 | 0.5704 | H-2→L | 30.3 | 5.1130 | 242.49 | 0.3887 | H→L+1 | 47.7 | ||
| 4.3488 | 285.10 | 0.4797 | H→L | 81.0 | 4.3673 | 283.89 | 0.4496 | H-1→L | 67.0 | ||
| 2 | 6.3847 | 194.19 | 0.3371 | H-5→L | 23.3 | 9 | 6.1917 | 200.24 | 0.5627 | H-2→L+1 | 30.9 |
| 5.3652 | 231.09 | 0.5613 | H-2→L | 30.2 | 5.1647 | 240.06 | 0.3556 | H→L+1 | 39.8 | ||
| 4.3338 | 286.08 | 0.4922 | H→L | 63.0 | 4.2466 | 291.96 | 0.6038 | H-1→L | 71.1 | ||
| 3 | 6.4021 | 193.66 | 0.4543 | H-3→L+1 | 55.8 | 10 | 6.1585 | 201.32 | 0.5873 | H-1→L+4 | 28.7 |
| 4.8064 | 257.96 | 0.3351 | H-2→L | 89.0 | 4.2134 | 294.26 | 0.4681 | H→L | 76.7 | ||
| 4 | 6.3085 | 196.54 | 0.5600 | H-1→L+4 | 31.9 | 11 | 6.3575 | 195.02 | 0.3613 | H-5→L | 22.3 |
| 4.8254 | 256.94 | 0.2768 | H-3→L | 83.7 | 5.3316 | 232.54 | 0.5412 | H→L+1 | 25.9 | ||
| 5 | 6.6481 | 186.50 | 0.4874 | H-3→L+2 | 27.7 | 4.3172 | 287.18 | 0.3724 | H→L | 81.2 | |
| 5.2108 | 237.94 | 0.5759 | H→L+1 | 23.0 | 12 | 6.3482 | 195.31 | 0.3164 | H-5→L | 34.4 | |
| 3.7785 | 328.13 | 0.6297 | H→L | 92.0 | 5.3256 | 232.81 | 0.5315 | H-1→L+1 | 36.0 | ||
| 6 | 6.0724 | 204.18 | 0.3920 | H-2→L+2 | 22.9 | 4.3063 | 287.91 | 0.3919 | H→L | 51.0 | |
| 5.2094 | 238.00 | 0.5679 | H→L+1 | 21.8 | 13 | 6.0731 | 204.15 | 0.5142 | H-1→L+4 | 26.4 | |
| 3.7664 | 329.18 | 0.6245 | H→L | 88.2 | 4.1614 | 297.94 | 0.5974 | H→L | 48.2 | ||
| 7 | 6.6384 | 186.77 | 0.3346 | H-3→L+3 | 24.5 | 14 | 6.2805 | 197.41 | 0.5505 | H-3→L+1 | 19.6 |
| 5.4868 | 225.97 | 0.4272 | H→L+2 | 29.4 | 4.7302 | 262.11 | 0.3216 | H-2→L | 53.8 | ||
| 3.7509 | 330.55 | 0.6480 | H→L | 86.7 | |||||||
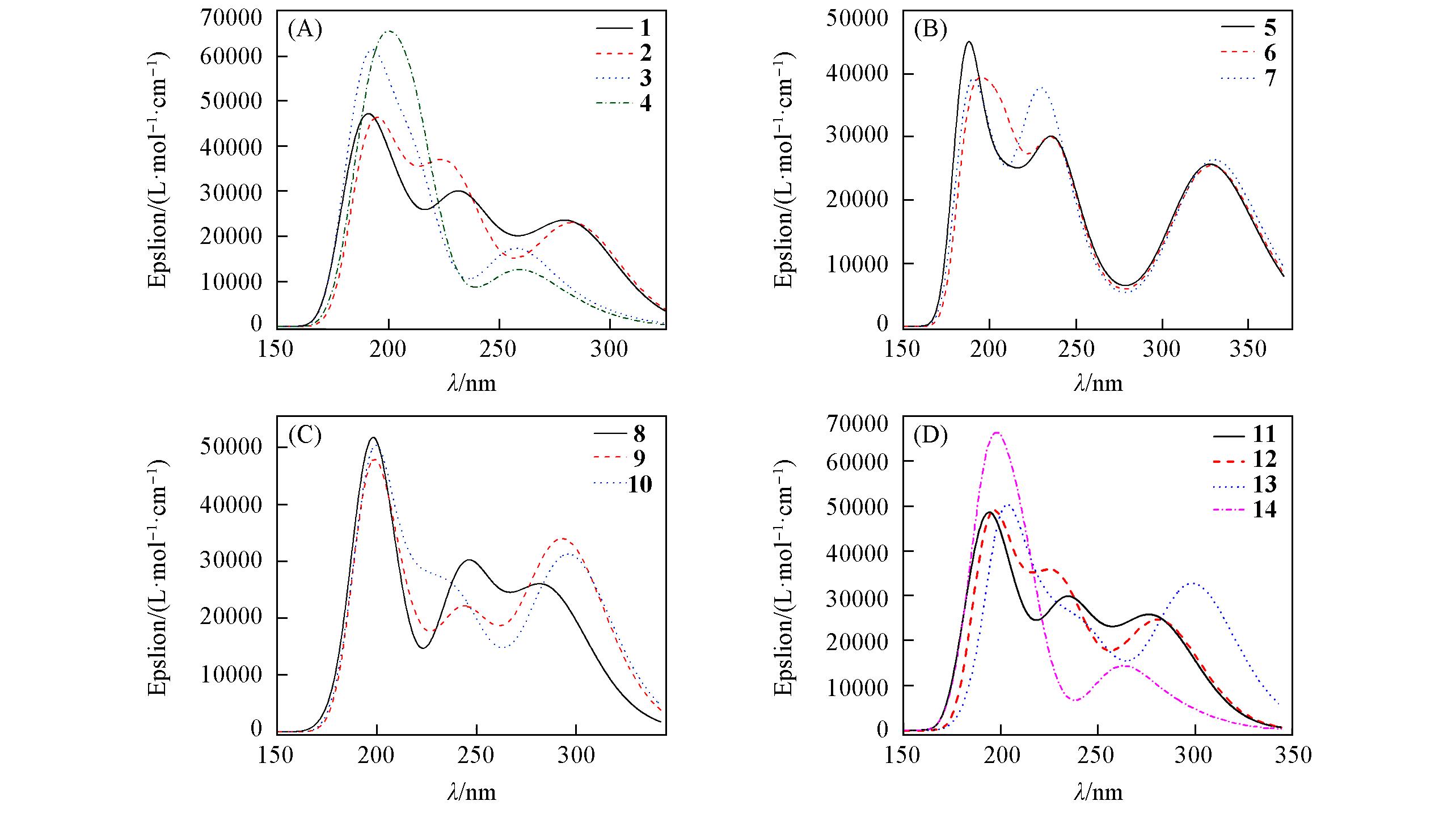
Fig.5 UV?Vis spectra of 14 flavonoids in methanol by ωB97XD/6?311+G(2d, p) method
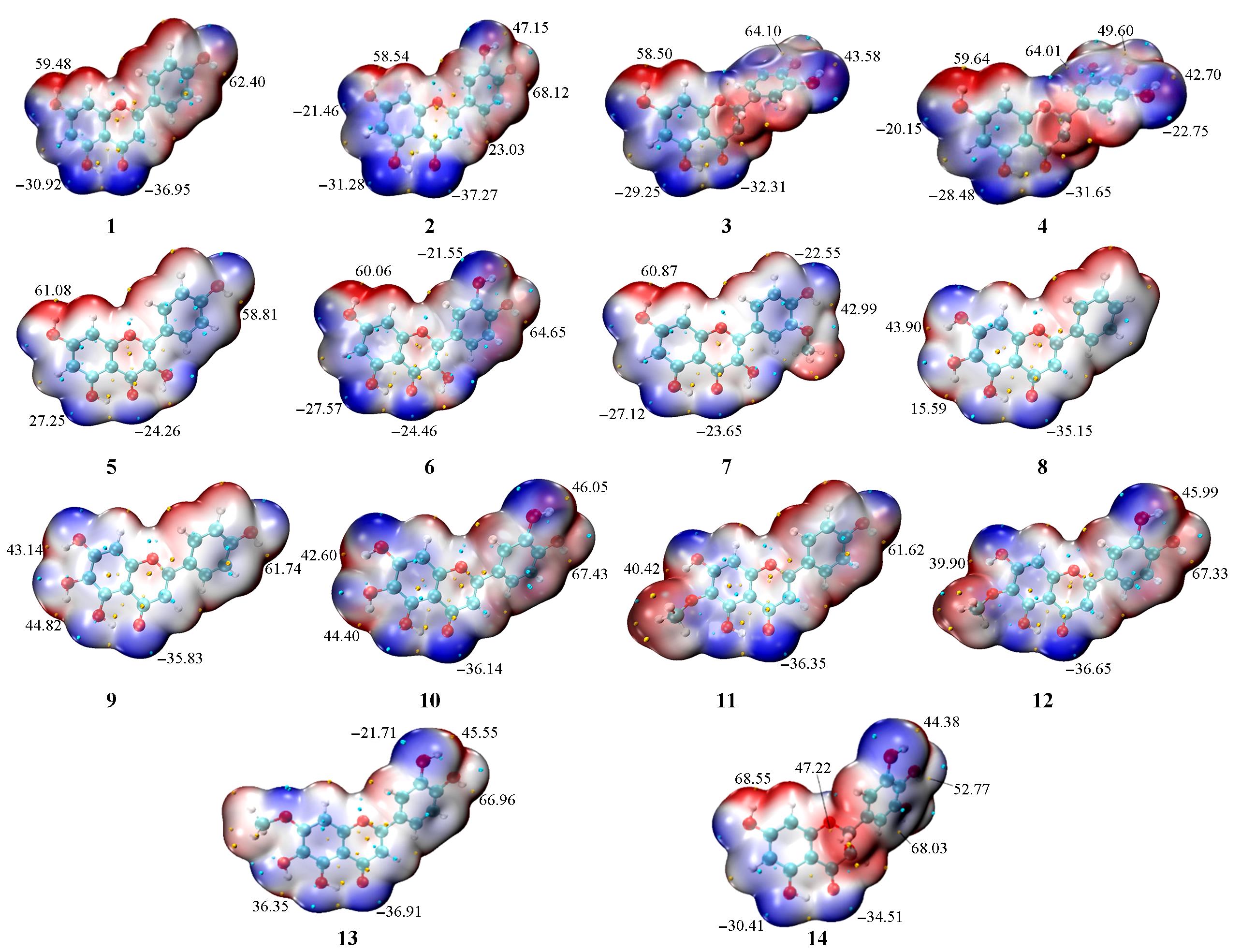
Fig.6 Molecular electrostatic potential maps of 14 flavonoids by ωB97XD/6?311+G(2d, p) method
| Compd. | HOMO | LUMO | Δε | IP | EA | χ | μ | η | S | ω |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | -8.2957 | -0.1405 | 8.1553 | 8.2957 | 0.1405 | 4.2181 | -4.2181 | 4.0776 | 0.2452 | 2.1817 |
| 2 | -8.2663 | -0.1309 | 8.1354 | 8.2663 | 0.1309 | 4.1986 | -4.1986 | 4.0677 | 0.2458 | 2.1669 |
| 3 | -8.2677 | 0.1083 | 8.3760 | 8.2677 | -0.1083 | 4.0797 | -4.0797 | 4.1880 | 0.2388 | 1.9871 |
| 4 | -8.2881 | -0.0199 | 8.2682 | 8.2881 | 0.0199 | 4.1540 | -4.1540 | 4.1341 | 0.2418 | 2.0870 |
| 5 | -8.0322 | -0.2760 | 7.7562 | 8.0322 | 0.2760 | 4.1541 | -4.1541 | 3.8781 | 0.2578 | 2.2249 |
| 6 | -8.0284 | -0.0291 | 7.9993 | 8.0284 | 0.0291 | 4.0288 | -4.0288 | 3.9996 | 0.2500 | 2.0291 |
| 7 | -7.9748 | -0.2940 | 7.6808 | 7.9748 | 0.2940 | 4.1344 | -4.1344 | 3.8404 | 0.2604 | 2.2254 |
| 8 | -7.9601 | -0.2864 | 7.6737 | 7.9601 | 0.2864 | 4.1232 | -4.1232 | 3.8369 | 0.2606 | 2.2155 |
| 9 | -7.9239 | -0.1682 | 7.7556 | 7.9239 | 0.1682 | 4.0461 | -4.0461 | 3.8778 | 0.2578 | 2.1108 |
| 10 | -7.9073 | -0.1606 | 7.7467 | 7.9073 | 0.1606 | 4.0339 | -4.0339 | 3.8733 | 0.2582 | 2.1006 |
| 11 | -8.1996 | -0.1563 | 8.0434 | 8.1996 | 0.1563 | 4.1779 | -4.1779 | 4.0217 | 0.2486 | 2.1701 |
| 12 | -8.1811 | -0.1465 | 8.0347 | 8.1811 | 0.1465 | 4.1638 | -4.1638 | 4.0173 | 0.2490 | 2.1578 |
| 13 | -7.9519 | -0.3038 | 7.6481 | 7.9519 | 0.3038 | 4.1279 | -4.1279 | 3.8241 | 0.2616 | 2.2279 |
| 14 | -8.3118 | -0.1064 | 8.2053 | 8.3118 | 0.1064 | 4.2091 | -4.2091 | 4.1027 | 0.2438 | 2.1592 |
Table 4 Global reactivity descriptors of 14 flavonoids by ωB97XD/6-311+G(2d, p) method
| Compd. | HOMO | LUMO | Δε | IP | EA | χ | μ | η | S | ω |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | -8.2957 | -0.1405 | 8.1553 | 8.2957 | 0.1405 | 4.2181 | -4.2181 | 4.0776 | 0.2452 | 2.1817 |
| 2 | -8.2663 | -0.1309 | 8.1354 | 8.2663 | 0.1309 | 4.1986 | -4.1986 | 4.0677 | 0.2458 | 2.1669 |
| 3 | -8.2677 | 0.1083 | 8.3760 | 8.2677 | -0.1083 | 4.0797 | -4.0797 | 4.1880 | 0.2388 | 1.9871 |
| 4 | -8.2881 | -0.0199 | 8.2682 | 8.2881 | 0.0199 | 4.1540 | -4.1540 | 4.1341 | 0.2418 | 2.0870 |
| 5 | -8.0322 | -0.2760 | 7.7562 | 8.0322 | 0.2760 | 4.1541 | -4.1541 | 3.8781 | 0.2578 | 2.2249 |
| 6 | -8.0284 | -0.0291 | 7.9993 | 8.0284 | 0.0291 | 4.0288 | -4.0288 | 3.9996 | 0.2500 | 2.0291 |
| 7 | -7.9748 | -0.2940 | 7.6808 | 7.9748 | 0.2940 | 4.1344 | -4.1344 | 3.8404 | 0.2604 | 2.2254 |
| 8 | -7.9601 | -0.2864 | 7.6737 | 7.9601 | 0.2864 | 4.1232 | -4.1232 | 3.8369 | 0.2606 | 2.2155 |
| 9 | -7.9239 | -0.1682 | 7.7556 | 7.9239 | 0.1682 | 4.0461 | -4.0461 | 3.8778 | 0.2578 | 2.1108 |
| 10 | -7.9073 | -0.1606 | 7.7467 | 7.9073 | 0.1606 | 4.0339 | -4.0339 | 3.8733 | 0.2582 | 2.1006 |
| 11 | -8.1996 | -0.1563 | 8.0434 | 8.1996 | 0.1563 | 4.1779 | -4.1779 | 4.0217 | 0.2486 | 2.1701 |
| 12 | -8.1811 | -0.1465 | 8.0347 | 8.1811 | 0.1465 | 4.1638 | -4.1638 | 4.0173 | 0.2490 | 2.1578 |
| 13 | -7.9519 | -0.3038 | 7.6481 | 7.9519 | 0.3038 | 4.1279 | -4.1279 | 3.8241 | 0.2616 | 2.2279 |
| 14 | -8.3118 | -0.1064 | 8.2053 | 8.3118 | 0.1064 | 4.2091 | -4.2091 | 4.1027 | 0.2438 | 2.1592 |
| Compd. | Atom | f- | f+ | f 0 | s- | s + | ω- | ω + |
|---|---|---|---|---|---|---|---|---|
| 1 | C7 | 1.1812 | 5.0235 | 3.1024 | 0.5298 | 2.2536 | 4.6870 | 19.9329 |
| O18 | 2.8332 | 1.4994 | 2.1663 | 1.2710 | 0.6726 | 11.2420 | 5.9495 | |
| 2 | C7 | 0.7940 | 4.7216 | 2.7578 | 0.7152 | 2.1266 | 3.1377 | 18.658 |
| O18 | 2.6189 | 1.4955 | 2.0572 | 1.1796 | 0.6736 | 10.3487 | 5.9096 | |
| 3 | C14 | 5.6469 | 3.7358 | 4.6913 | 1.3496 | 0.8928 | 11.2209 | 7.4234 |
| O17 | 0.4101 | 5.4176 | 2.9139 | 0.0980 | 1.2948 | 0.8150 | 10.7653 | |
| 4 | C4 | 6.1983 | 1.6941 | 3.9462 | 1.4988 | 0.4096 | 12.9358 | 3.5357 |
| O17 | 3.5260 | 5.3024 | 4.4142 | 0.4263 | 1.2822 | 7.3588 | 11.0660 | |
| 5 | C13 | -7.1566 | 6.7652 | -0.1957 | -1.8450 | 1.7440 | -15.9226 | 15.0519 |
| O19 | 3.9242 | 1.3889 | 2.6565 | 1.0116 | 0.3580 | 8.7310 | 3.0901 | |
| 6 | C14 | 1.2296 | 17.3725 | 9.3011 | 0.5726 | 8.0886 | 4.6575 | 65.8019 |
| O18 | 3.1287 | 1.8626 | 2.4956 | 1.4568 | 0.8672 | 11.8504 | 7.0551 | |
| 7 | C13 | -6.7374 | 4.9070 | -0.9152 | -1.7544 | 1.2778 | -14.9935 | 10.9200 |
| O21 | 5.5212 | -2.4698 | 1.5257 | 1.4378 | -0.6432 | 12.2868 | -5.4963 | |
| 8 | C7 | 0.6094 | 5.7505 | 3.1800 | 0.3044 | 2.8730 | 2.5712 | 24.2639 |
| O20 | 3.7757 | 1.1446 | 2.4601 | 1.8864 | 0.5718 | 15.9311 | 4.8294 | |
| 9 | C7 | -0.0812 | 5.0206 | 2.4697 | -0.0398 | 2.4610 | -0.3239 | 20.0216 |
| O21 | 3.5850 | 1.1511 | 2.3681 | 1.7574 | 0.5642 | 14.2965 | 4.5906 | |
| 10 | C14 | 2.6090 | 5.6722 | 4.1406 | 1.2820 | 2.7874 | 10.3675 | 22.5401 |
| O20 | 2.8740 | 1.1491 | 2.0115 | 1.4122 | 0.5646 | 11.4205 | 4.5663 | |
| 11 | C7 | 0.6740 | 4.4612 | 2.5676 | 0.3102 | 2.0540 | 2.6839 | 17.7653 |
| O21 | 2.3575 | 0.8734 | 1.6155 | 1.0854 | 0.4022 | 9.3882 | 3.4780 | |
| 12 | C14 | 2.9389 | 5.7603 | 4.3496 | 1.3560 | 2.6578 | 11.6547 | 22.8437 |
| O21 | 2.1916 | 0.8821 | 1.5368 | 1.0112 | 0.4070 | 8.6911 | 3.4980 | |
| 13 | C7 | 0.0819 | 5.6552 | 2.8686 | 0.0314 | 1.4794 | 0.1825 | 12.5993 |
| O20 | 3.5217 | 0.8879 | 2.2048 | 0.9212 | 0.2322 | 7.8460 | 1.9782 | |
| 14 | C15 | 9.5655 | -16.8933 | -3.6639 | 2.3320 | -4.1186 | 20.6539 | -36.4761 |
| C16 | -8.2259 | 19.4100 | 5.5920 | -2.0054 | 4.7322 | -17.7613 | 41.9100 |
Table 5 Condensed Fukui function values of 14 flavonoids by ωB97XD/6-311+G(2d, p) method
| Compd. | Atom | f- | f+ | f 0 | s- | s + | ω- | ω + |
|---|---|---|---|---|---|---|---|---|
| 1 | C7 | 1.1812 | 5.0235 | 3.1024 | 0.5298 | 2.2536 | 4.6870 | 19.9329 |
| O18 | 2.8332 | 1.4994 | 2.1663 | 1.2710 | 0.6726 | 11.2420 | 5.9495 | |
| 2 | C7 | 0.7940 | 4.7216 | 2.7578 | 0.7152 | 2.1266 | 3.1377 | 18.658 |
| O18 | 2.6189 | 1.4955 | 2.0572 | 1.1796 | 0.6736 | 10.3487 | 5.9096 | |
| 3 | C14 | 5.6469 | 3.7358 | 4.6913 | 1.3496 | 0.8928 | 11.2209 | 7.4234 |
| O17 | 0.4101 | 5.4176 | 2.9139 | 0.0980 | 1.2948 | 0.8150 | 10.7653 | |
| 4 | C4 | 6.1983 | 1.6941 | 3.9462 | 1.4988 | 0.4096 | 12.9358 | 3.5357 |
| O17 | 3.5260 | 5.3024 | 4.4142 | 0.4263 | 1.2822 | 7.3588 | 11.0660 | |
| 5 | C13 | -7.1566 | 6.7652 | -0.1957 | -1.8450 | 1.7440 | -15.9226 | 15.0519 |
| O19 | 3.9242 | 1.3889 | 2.6565 | 1.0116 | 0.3580 | 8.7310 | 3.0901 | |
| 6 | C14 | 1.2296 | 17.3725 | 9.3011 | 0.5726 | 8.0886 | 4.6575 | 65.8019 |
| O18 | 3.1287 | 1.8626 | 2.4956 | 1.4568 | 0.8672 | 11.8504 | 7.0551 | |
| 7 | C13 | -6.7374 | 4.9070 | -0.9152 | -1.7544 | 1.2778 | -14.9935 | 10.9200 |
| O21 | 5.5212 | -2.4698 | 1.5257 | 1.4378 | -0.6432 | 12.2868 | -5.4963 | |
| 8 | C7 | 0.6094 | 5.7505 | 3.1800 | 0.3044 | 2.8730 | 2.5712 | 24.2639 |
| O20 | 3.7757 | 1.1446 | 2.4601 | 1.8864 | 0.5718 | 15.9311 | 4.8294 | |
| 9 | C7 | -0.0812 | 5.0206 | 2.4697 | -0.0398 | 2.4610 | -0.3239 | 20.0216 |
| O21 | 3.5850 | 1.1511 | 2.3681 | 1.7574 | 0.5642 | 14.2965 | 4.5906 | |
| 10 | C14 | 2.6090 | 5.6722 | 4.1406 | 1.2820 | 2.7874 | 10.3675 | 22.5401 |
| O20 | 2.8740 | 1.1491 | 2.0115 | 1.4122 | 0.5646 | 11.4205 | 4.5663 | |
| 11 | C7 | 0.6740 | 4.4612 | 2.5676 | 0.3102 | 2.0540 | 2.6839 | 17.7653 |
| O21 | 2.3575 | 0.8734 | 1.6155 | 1.0854 | 0.4022 | 9.3882 | 3.4780 | |
| 12 | C14 | 2.9389 | 5.7603 | 4.3496 | 1.3560 | 2.6578 | 11.6547 | 22.8437 |
| O21 | 2.1916 | 0.8821 | 1.5368 | 1.0112 | 0.4070 | 8.6911 | 3.4980 | |
| 13 | C7 | 0.0819 | 5.6552 | 2.8686 | 0.0314 | 1.4794 | 0.1825 | 12.5993 |
| O20 | 3.5217 | 0.8879 | 2.2048 | 0.9212 | 0.2322 | 7.8460 | 1.9782 | |
| 14 | C15 | 9.5655 | -16.8933 | -3.6639 | 2.3320 | -4.1186 | 20.6539 | -36.4761 |
| C16 | -8.2259 | 19.4100 | 5.5920 | -2.0054 | 4.7322 | -17.7613 | 41.9100 |
| Compd. | Mw | HBD | HBA | TPSA | Log(BCF) | ST | Density | Polarizability | lgP | Lipinski | Solubility |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 270.24 | 3 | 5 | 86.99 | 1.37 | 79.60 | 1.55 | 27.69 | 2.45 | Good | Soluble |
| 2 | 286.24 | 4 | 6 | 107.22 | 1.59 | 92.60 | 1.65 | 28.44 | 2.35 | Good | Soluble |
| 3 | 288.25 | 4 | 6 | 107.22 | 1.65 | 84.38 | 1.59 | 28.6 | 2.35 | Good | Soluble |
| 4 | 304.25 | 5 | 7 | 127.45 | 1.29 | 97.42 | 1.69 | 29.34 | 1.98 | Moderate | Soluble |
| 5 | 286.24 | 4 | 6 | 107.22 | 1.33 | 98.93 | 1.69 | 28.32 | 2.12 | Good | Soluble |
| 6 | 302.24 | 5 | 7 | 127.45 | 1.35 | 114.88 | 1.80 | 29.07 | 1.95 | Good | Soluble |
| 7 | 316.26 | 4 | 7 | 116.45 | 1.11 | 88.31 | 1.63 | 30.97 | 2.10 | Good | Soluble |
| 8 | 270.24 | 3 | 5 | 86.99 | 2.29 | 79.60 | 1.55 | 27.69 | 3.03 | Good | Soluble |
| 9 | 286.24 | 4 | 6 | 107.22 | 1.70 | 92.60 | 1.65 | 28.44 | 2.41 | Good | Soluble |
| 10 | 302.24 | 5 | 7 | 127.45 | 1.92 | 107.40 | 1.76 | 29.18 | 2.34 | Good | Soluble |
| 11 | 300.26 | 3 | 6 | 96.22 | 0.98 | 72.81 | 1.51 | 30.34 | 2.55 | Good | Insoluble |
| 12 | 316.26 | 4 | 7 | 116.45 | 1.21 | 83.36 | 1.61 | 31.08 | 2.2 | Good | Soluble |
| 13 | 316.26 | 4 | 7 | 116.45 | 1.85 | 83.36 | 1.61 | 31.08 | 2.35 | Good | Soluble |
| 14 | 320.25 | 6 | 8 | 147.68 | 0.71 | 115.7 | 1.81 | 29.97 | 1.31 | Moderate | Soluble |
Table 6 Drug-like evaluation of 14 flavonoids*
| Compd. | Mw | HBD | HBA | TPSA | Log(BCF) | ST | Density | Polarizability | lgP | Lipinski | Solubility |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 270.24 | 3 | 5 | 86.99 | 1.37 | 79.60 | 1.55 | 27.69 | 2.45 | Good | Soluble |
| 2 | 286.24 | 4 | 6 | 107.22 | 1.59 | 92.60 | 1.65 | 28.44 | 2.35 | Good | Soluble |
| 3 | 288.25 | 4 | 6 | 107.22 | 1.65 | 84.38 | 1.59 | 28.6 | 2.35 | Good | Soluble |
| 4 | 304.25 | 5 | 7 | 127.45 | 1.29 | 97.42 | 1.69 | 29.34 | 1.98 | Moderate | Soluble |
| 5 | 286.24 | 4 | 6 | 107.22 | 1.33 | 98.93 | 1.69 | 28.32 | 2.12 | Good | Soluble |
| 6 | 302.24 | 5 | 7 | 127.45 | 1.35 | 114.88 | 1.80 | 29.07 | 1.95 | Good | Soluble |
| 7 | 316.26 | 4 | 7 | 116.45 | 1.11 | 88.31 | 1.63 | 30.97 | 2.10 | Good | Soluble |
| 8 | 270.24 | 3 | 5 | 86.99 | 2.29 | 79.60 | 1.55 | 27.69 | 3.03 | Good | Soluble |
| 9 | 286.24 | 4 | 6 | 107.22 | 1.70 | 92.60 | 1.65 | 28.44 | 2.41 | Good | Soluble |
| 10 | 302.24 | 5 | 7 | 127.45 | 1.92 | 107.40 | 1.76 | 29.18 | 2.34 | Good | Soluble |
| 11 | 300.26 | 3 | 6 | 96.22 | 0.98 | 72.81 | 1.51 | 30.34 | 2.55 | Good | Insoluble |
| 12 | 316.26 | 4 | 7 | 116.45 | 1.21 | 83.36 | 1.61 | 31.08 | 2.2 | Good | Soluble |
| 13 | 316.26 | 4 | 7 | 116.45 | 1.85 | 83.36 | 1.61 | 31.08 | 2.35 | Good | Soluble |
| 14 | 320.25 | 6 | 8 | 147.68 | 0.71 | 115.7 | 1.81 | 29.97 | 1.31 | Moderate | Soluble |
| Compd. | P?gp Substrates | CYP1A2 Inhibitor | CYP2C9 Inhibitor | CYP2C19 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Ames | hERG | Caco?2 | PPB | CNS | HIA | Metabolic Stability |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NS | EI | I | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 2 | NS | EI | I | U | NI | U | U | U | MP | EB | NP | HA | U |
| 3 | NS | EI | I | U | NI | U | U | U | MP | EB | NP | HA | U |
| 4 | NS | U | U | U | NI | NI | M | NI | MP | SB | NP | HA | S |
| 5 | NS | I | U | U | NI | U | M | NI | MP | SB | NP | HA | U |
| 6 | NS | EI | U | U | NI | I | U | NI | MP | EB | NP | HA | U |
| 7 | NS | EI | I | U | NI | U | U | NI | MP | EB | NP | HA | U |
| 8 | NS | EI | U | U | NI | I | M | NI | HP | SB | NP | HA | U |
| 9 | NS | EI | U | I | NI | U | M | NI | HP | EB | WP | HA | U |
| 10 | NS | EI | I | U | U | U | M | NI | MP | EB | NP | HA | U |
| 11 | NS | EI | U | I | NI | U | U | NI | HP | EB | NP | HA | U |
| 12 | NS | EI | I | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 13 | NS | EI | U | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 14 | NS | U | U | U | NI | NI | M | NI | MP | EB | NP | HA | S |
Table 7 The pharmacokinetic parameters of 14 flavonoids*
| Compd. | P?gp Substrates | CYP1A2 Inhibitor | CYP2C9 Inhibitor | CYP2C19 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Ames | hERG | Caco?2 | PPB | CNS | HIA | Metabolic Stability |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | NS | EI | I | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 2 | NS | EI | I | U | NI | U | U | U | MP | EB | NP | HA | U |
| 3 | NS | EI | I | U | NI | U | U | U | MP | EB | NP | HA | U |
| 4 | NS | U | U | U | NI | NI | M | NI | MP | SB | NP | HA | S |
| 5 | NS | I | U | U | NI | U | M | NI | MP | SB | NP | HA | U |
| 6 | NS | EI | U | U | NI | I | U | NI | MP | EB | NP | HA | U |
| 7 | NS | EI | I | U | NI | U | U | NI | MP | EB | NP | HA | U |
| 8 | NS | EI | U | U | NI | I | M | NI | HP | SB | NP | HA | U |
| 9 | NS | EI | U | I | NI | U | M | NI | HP | EB | WP | HA | U |
| 10 | NS | EI | I | U | U | U | M | NI | MP | EB | NP | HA | U |
| 11 | NS | EI | U | I | NI | U | U | NI | HP | EB | NP | HA | U |
| 12 | NS | EI | I | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 13 | NS | EI | U | I | NI | I | U | NI | HP | EB | NP | HA | U |
| 14 | NS | U | U | U | NI | NI | M | NI | MP | EB | NP | HA | S |
| 1 | National Pharmacopoeia Commission, Pharmacopoeia of the People’s Republic of China, China Medical Science and Technology Press, Beijing, 2020, 70—71(国家药典委员会. 中华人民共和国药典, 北京: 中国医药科技出版社, 2020, 70—71) |
| 2 | Li L., Liu Z. Q., Liu C. M., Tsao R., Lv L., Liu S. Y., Chem. J. Chinese Universities, 2006, 8(27), 1430—1434(李丽, 刘志强, 刘春明, Tsao R., 吕磊, 刘淑莹. 高等学校化学学报, 2006, 8(27), 1430—1434) |
| 3 | Wu F. H, Liang J. Y., Chen R., Wang Q. Z., Li W. G., Chin. J. Nat. Medicines, 2006,4(6), 435—439(吴斐华, 梁敬钰, 陈荣, 王奇志, 李伟光. 中国天然药物, 2006, 4(6), 435—439) |
| 4 | Wu F. H., Liang J. Y., Yu P., Cai S. F., Chin. J. Chin. Mater. Med., 2005, 30(15), 1179—1183(吴斐华, 梁敬钰, 余平, 蔡松福. 中国中药杂志, 2005, 30(15), 1179—1183) |
| 5 | Qian Y., Bai H. B., Hu R., Chin. J. Modern Appl. Pharm., 2011, 28(5), 406—408(钱莺, 白海波, 扈荣. 中国现代应用药学, 2011, 28(5), 406—408) |
| 6 | Wang Q., Tao H. Q., World Latest Medicine Information, 2017, 17(A2), 72—75(王琪, 陶河清. 世界最新医学信息文摘, 2017, 17(A2), 72—75) |
| 7 | Xia L. H., Jin G. Q., Sun L., Yang J., China Pharm., 2013, 16(2), 294—296(夏玲红, 金冠钦, 孙黎, 杨娟. 中国药师, 2013, 16(2), 294—296) |
| 8 | Yan P. F., Liu G. Y., Zhao S. M., Song L. L., Tan L. N., Jin Y. R., Li X. W., China Pharm. J., 2009, 44(1), 19—21(颜佩芳, 刘桂英, 赵士敏, 宋丽丽, 谭丽娜, 金永日, 李绪文. 中国药学杂志, 2009, 44(1), 19—21) |
| 9 | Liu Y. H., Zhao Y., Journal of Hunan Institute of Science and Technology, 2009, 22(2), 63—64(刘艳辉, 赵映. 湖南理工学院学报, 2009, 22(2), 63—64) |
| 10 | Yan Q. P., Protective Effects of Extracts of Plantain and Eucommia Ulmoides on Liver Injury, Guangdong University of Technology, Guangzhou, 2012(颜秋萍. 车前草和杜仲提取物对肝损伤的保护作用, 广州: 广东工业大学, 2012) |
| 11 | Zhang X. Q., Qu W., Liang J. Y., Strait Pharm. J., 2013, 25(11), 1—8(张雪芹, 曲玮, 梁敬钰. 海峡药学, 2013, 25(11), 1—8) |
| 12 | Giulia D. C., Nicola M., Angelo A., Life Sci., 1999, 65(4), 337—353 |
| 13 | Nishibe S., J. Nat. Med. Tokyo, 1997, 51(6), 547—549 |
| 14 | Wang Y. H., Fang Y. M., Tan P., Zhang C. S., Guizhou Agric. Sci., 2010, 38(8), 50—52(王毅红, 方玉梅, 谭萍, 张春生. 贵州农业科学, 2010, 38(8), 50—52) |
| 15 | Xia D. Z., Liu J. E., Chen P. P., Bull. Sci. Technol., 2009, 25(6), 792—797(夏道宗, 刘杰尔, 陈佩佩. 科技通报, 2009, 25(6), 792—797) |
| 16 | Roginsky V. A., Barsukova T. K., Bors W., J. Am. Oil. Chem. Soc., 1996, 73(6), 6—10 |
| 17 | Salzner U., Aydin A., J. Chem. Theory Comput., 2011, 7(8), 2568—2583 |
| 18 | Holtomo O., Motapon O., Nsangou M., J. Phys. Chem. A, 2019, 123(48), 10437—10445 |
| 19 | Santiago P. H. O., Tiago F. S., Castro M. S., Souza P. E. N., Martins J. B. L., Gatto C. C., J. Inorg. Biochem., 2020, 204, 110949 |
| 20 | Sun Y., Midas T., Zhou W. J., Lu W. C., Liu J. B., J. Phys. Chem. B, 2019, 123(49), 10410—10423 |
| 21 | Miertuš S., Tomasi J., J. Chem. Phy., 1982, 65(2), 239—245 |
| 22 | Bi T. J., Wang F., Wang Z. F., Chem. J. Chinese Universities, 2020, 41(12), 2781—2787(毕婷君, 王繁, 王治钒. 高等学校化学学报, 2020, 41(12), 2781—2787) |
| 23 | Parr R., Yang W., Density⁃Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989,70—86 |
| 24 | Geerlings P., De Proft F., Langenaeker W., Chem. Rev., 2003, 103(5), 1793—1873 |
| 25 | Fu R., Lu T., Chen F. W., Acta. Phys. Chim. Sin.., 2014, 30(4), 628—639(付蓉, 卢天, 陈飞武. 物理化学学报, 2014, 30(4), 628—639) |
| 26 | Frisch M. J., Trucks G. W., Schlegel H. B., Gaussian 16, Revision B.01, Gaussian Inc, Wallingford CT, 2016 |
| 27 | Lu T., Chen F., J. Comput. Chem., 2012, 33(5), 580—592 |
| 28 | Humphrey W., Dalke A., Schulten K., J. Molec. Graphics, 1996, 14(1), 33—38 |
| 29 | Lv Z. B., Zhuang L. H., Tian Y. W., Wang D. L., J. Mole. Sci., 2007, 23(5), 354—357(吕振波, 庄丽宏, 田彦文, 王东来. 分子科学学报, 2007, 23(5), 354—357) |
| 30 | Lu R. M., Li Y. Q., Wang X., Zhang J. Y., Li B., Wei J. H., Journal of Guangxi Normal University(Natural Science Edition), 2020, 38(5), 78—85(卢汝梅, 黎云清, 王肖, 张进燕, 李兵, 韦建华. 广西师范大学学报(自然科学版), 2020, 38(5), 78—85) |
| 31 | Peng G., Huang J., Zhou Y. J., Chou A., Peng D., China Tradit. Herb. Drugs, 2020, 51(19), 4902—4906(彭谷, 黄娟, 周应军, 丑安, 彭电. 中草药, 2020, 51(19), 4902—4906) |
| 32 | Gao M. Z., Zhang H., Lin L., Xu Q., Qin F., Yuan C. L., J. Liaoning Univ. Tradit. Chin. Med., 2003, 5(2), 157(高明哲, 张惠, 林立, 徐青, 覃芳, 袁昌鲁. 辽宁中医学院学报, 2003, 5(2), 157) |
| 33 | Wang S. S., Huang W. Z., Zeng G. Z., Li G. P., Zhu H., Zhang Z., Gao X. M., Journal of Yunnan Nationalities University(Natural Science Edition), 2019, 28(2), 105—108(王闪闪, 黄文忠, 曾广智, 李干鹏, 朱鸿, 张再, 高雪梅. 云南民族大学学报(自然科学版), 2019, 28(2), 105—108) |
| 34 | Yi Y., Wu X., Wang Y., Ye W. C., Zhang Q. W., J. Chin. Med. Mater., 2011, 34(5), 712—716(易衍, 巫鑫, 王英, 叶文才, 张庆文. 中药材, 2011, 34(5), 712—716) |
| 35 | Ni F. Y., Wen J. H., Li M., Zhao Y. W., Wang Z. Z., Xiao W., China Tradit. Herb. Drugs, 2017, 48(18), 3689—3692(倪付勇, 温建辉, 李明, 赵祎武, 王振中, 萧伟. 中草药, 2017, 48(18), 3689—3692) |
| 36 | Duan X. H., Zhang X. W., Qin M., He P., Wang L. L., Zhao J. C., Pei L., Chen Y. L., Li C. H., China Tradit. Herb. Drugs, 2020, 51(15), 3863—3868(段绪红, 张学文, 秦梦, 何培, 王丽丽, 赵建成, 裴林, 陈玉玲, 李春花. 中草药, 2020, 51(15), 3863— 3868) |
| 37 | Fatma A. M., Mohamed S. M., Siham M. E., Ahmed H. G., Wafaa M. E., J. Pharm Pharmacol, 2012, 64(11), 1678—1687 |
| 38 | Qu C., Le S. J., Lin H., Kai J., Shang G. X., Tang Y. P., Tao W. W., Duan J. A., China Tradit. Herb. Drugs, 2015, 46(13), 1872—1877(瞿城, 乐世俊, 林航, 开均, 尚冠雄, 唐于平, 陶伟伟, 段金廒. 中草药, 2015, 46(13), 1872—1877) |
| 39 | Chao S. W., Su M. Y., Chiou L. C., Chen L. C., Chang C. I., Huang W. J., J. Nat. Prod., 2015, 78(8), 1969—1976 |
| 40 | Wen J. H., Ni F. Y., Li M., Xie X., Wang Y. X., Wu Y., Wang Z. Z., Xiao W., Chin. Pat. Med., 2019, 41(10), 2393—2397(温建辉, 倪付勇, 李 明, 谢雪, 王永香, 吴云, 王振中, 肖伟. 中成药, 2019, 41(10), 2393—2397) |
| 41 | Sheyla R., Olman H., Mikel G. I. C., Iciar A., Diana A., Rita Y. C., María I. C., LWT⁃Food Sci. Technol., 2010, 44(4), 875— 882 |
| 42 | Zheng S. J., Sun J. Y., Wu Y., Chen L. J., Li H., J. Sichuan Univ.(Engineering Science Edition), 2016, 48(1), 215—220(郑守军, 孙锦玉, 吴 悠, 陈俐娟, 李晖. 四川大学学报(工程科学版), 2016, 48(1), 215—220) |
| 43 | Jacquemin D., Preat J., Perpete E. A., Chem. Phys. Lett., 2005, 410(4—6), 254—259 |
| 44 | Lin L. D., Qin G. W., Xu R. S., Acta. Bot. Sin.,1994, 36(5), 393—397(林立东, 秦国伟, 徐任生. 植物学报, 1994, 36(5), 393—397) |
| 45 | Ding X. L., Wu J. M., Xu X., Chem. J. Chinese Universites, 2008, 29(2), 396—398(丁秀丽, 吴剑鸣, 徐昕. 高等学校化学学报, 2008, 29(2), 396—398) |
| 46 | Lipinski C. A., J. Pharmacol.Tox. Met., 2000, 44(1), 235—249 |
| 47 | Guo Z. R., Acta Pharm. Sin., 2010, 45(5), 539—547(郭宗儒. 药学学报, 2010, 45(5), 539—547) |
| 48 | Li Y., Zhu C. Y., Journal of Liaocheng University, 2019, 32(4), 1—5(黎迎, 朱春燕. 聊城大学学报, 2019, 32(4), 1—5) |
| 49 | Zhang W. D., Sun H., Song S. S., Dai G. L., Bai Y. T., Ju W. Z., New Chinese Medicines and Clinical Pharmacology, 2019, 30(6), 695—699(张卫东, 孙红, 宋珊珊, 戴国梁, 白永涛, 居文政. 中药新药与临床药理, 2019,30(6), 695—699) |
| 50 | Decleves X., Jacob A., Yousif S., Curr. Drug Metab., 2011, 12(8), 732—741 |
| 51 | Elmeliegy M., Vourvahis M., Guo C. C., Wang D. D., Clini. Pharmacokinet, 2020, 59(6), 699—714 |
| 52 | Gao Y., Yao B. Y., Zhou Z. C., Journal of Toxicology, 2016, 30(5), 334—338(高雅, 姚碧云, 周宗灿. 毒理学杂志, 2016, 30(5), 334—338) |
| [1] | 何鸿锐, 夏文生, 张庆红, 万惠霖. 羟基氧化铟团簇与二氧化碳和甲烷作用的密度泛函理论研究[J]. 高等学校化学学报, 2022, 43(8): 20220196. |
| [2] | 黄汉浩, 卢湫阳, 孙明子, 黄勃龙. 石墨炔原子催化剂的崭新道路:基于自验证机器学习方法的筛选策略[J]. 高等学校化学学报, 2022, 43(5): 20220042. |
| [3] | 刘洋, 李旺昌, 张竹霞, 王芳, 杨文静, 郭臻, 崔鹏. Sc3C2@C80与[12]CPP纳米环之间非共价相互作用的理论研究[J]. 高等学校化学学报, 2022, 43(11): 20220457. |
| [4] | 周成思, 赵远进, 韩美晨, 杨霞, 刘晨光, 贺爱华. 硅烷类外给电子体对丙烯-丁烯序贯聚合的调控作用[J]. 高等学校化学学报, 2022, 43(10): 20220290. |
| [5] | 王园月, 安梭梭, 郑旭明, 赵彦英. 5-巯基-1, 3, 4-噻二唑-2-硫酮微溶剂团簇的光谱和理论计算研究[J]. 高等学校化学学报, 2022, 43(10): 20220354. |
| [6] | 程媛媛, 郗碧莹. ·OH自由基引发CH3SSC |
| [7] | 马丽娟, 高升启, 荣祎斐, 贾建峰, 武海顺. Sc, Ti, V修饰B/N掺杂单缺陷石墨烯的储氢研究[J]. 高等学校化学学报, 2021, 42(9): 2842. |
| [8] | 钟声广, 夏文生, 张庆红, 万惠霖. 电中性团簇MCu2Ox(M=Cu2+, Ce4+, Zr4+)上甲烷和二氧化碳直接合成乙酸的理论研究[J]. 高等学校化学学报, 2021, 42(9): 2878. |
| [9] | 胡皓程, 李文利, 张嘉宁, 刘宇博. 黑木耳寡糖的提取、 结构表征及生物活性[J]. 高等学校化学学报, 2021, 42(8): 2465. |
| [10] | 郑若昕, 张颖, 徐昕. 低标度XYG3双杂化密度泛函的开发与测评[J]. 高等学校化学学报, 2021, 42(7): 2210. |
| [11] | 柳扬, 李清波, 孙杰, 赵显. Ga对在AlN衬底上直接生长石墨烯的远程催化[J]. 高等学校化学学报, 2021, 42(7): 2271. |
| [12] | 应富鸣, 计辰儒, 苏培峰, 吴玮. 基于完全活性空间自洽场的杂化多组态密度泛函方法λ-DFCAS[J]. 高等学校化学学报, 2021, 42(7): 2218. |
| [13] | 杨一莹, 朱荣秀, 张冬菊, 刘成卜. 金催化炔基苯并二𫫇英环化合成8-羟基异香豆素的理论研究[J]. 高等学校化学学报, 2021, 42(7): 2299. |
| [14] | 王建, 张红星. 四配位铂磷光发射体结构与光物理性质关系的理论研究[J]. 高等学校化学学报, 2021, 42(7): 2245. |
| [15] | 胡伟, 刘小峰, 李震宇, 杨金龙. 金刚石纳米线氮空位色心的表面与尺寸效应[J]. 高等学校化学学报, 2021, 42(7): 2178. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||