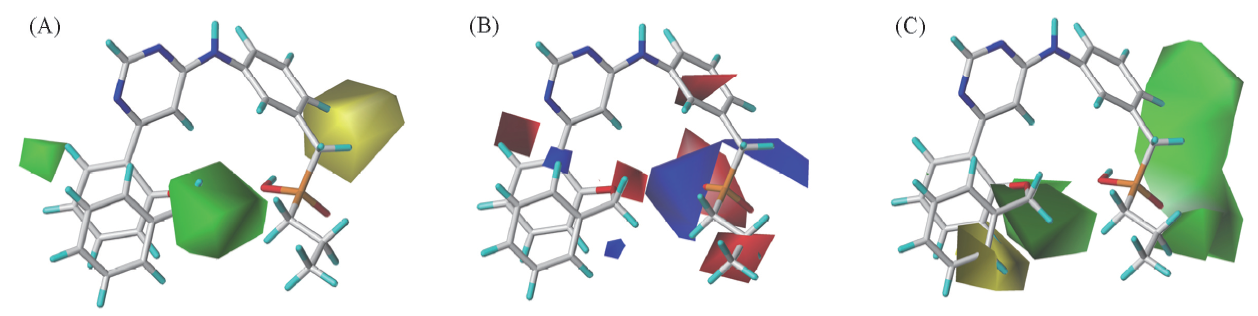
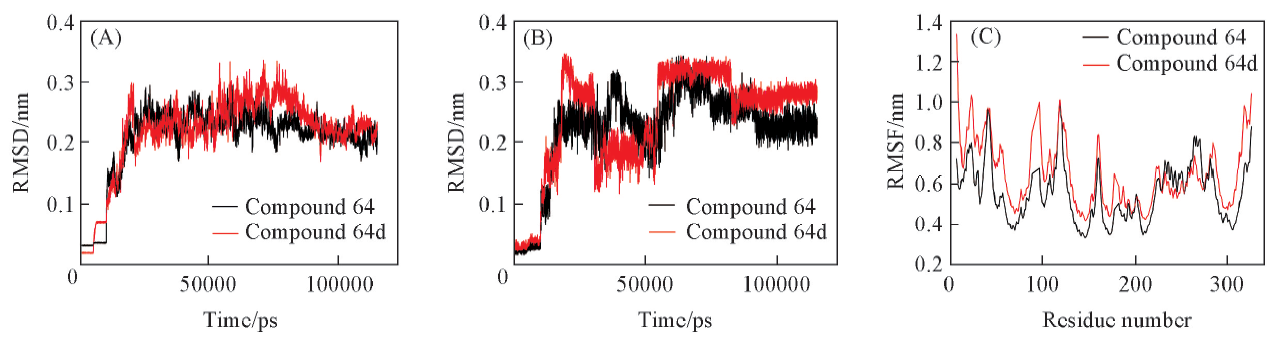
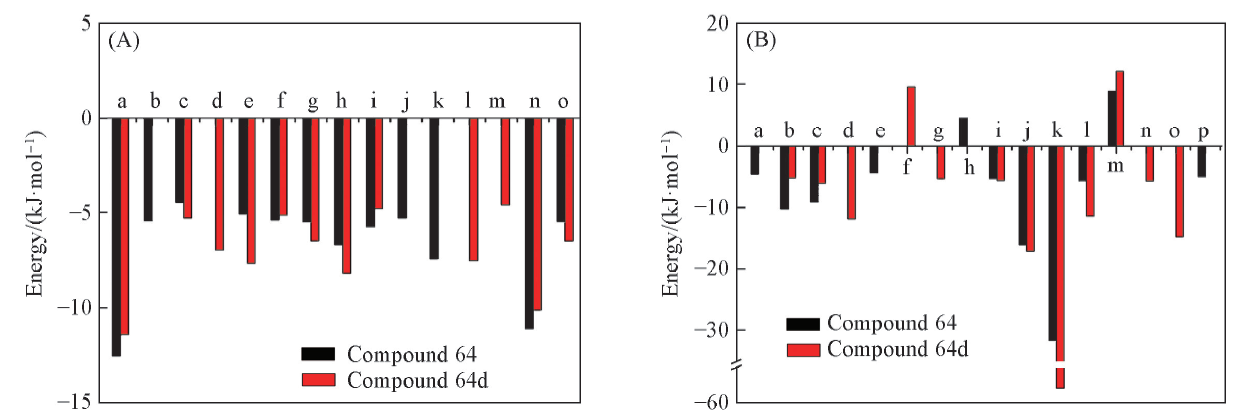
| [1] |
Cicenas J., Kalyan K., Sorokinas A., Jatulyte A., Valiunas D., Kaupinis A., Valius M., Cancers,2014, 6, 2224-2234
|
| [2] |
Asghar U., Witkiewicz A. K., Turner N. C., Knudsen E. S., Chem. J. Chinese Universities,2010, 31, 130-146
|
| [3] |
Lim S., Kaldis P., Development,2013, 140, 3079-3087
|
| [4] |
Liao Y., Feng Y., Shen J., Hornicek F. J., Duan Z., Cancer and Chem. J. Chinese Universities,2010, 31, 151-163
|
| [5] |
Kim M. S., Ji H. K., Jin M. K., Hong S. H., Lee K., Choi H. S., Sang M. W., Chang J. Y., Tai Y. K., Ko S. G. K., Chem. J. Chinese Universities,2010, 31, 3522-3522
|
| [6] |
Wang S., Fischer P. M., Chem. J. Chinese Universities,2010, 31, 302-313
|
| [7] |
Barrière C., Santamaría D., Cerqueira A., Galán J., Martín A., Ortega S., Malumbres M., Dubus P., Barbacid M., Chem. J. Chinese Universities,2010, 31, 72-81
|
| [8] |
Berthet C., Aleem E., Coppola V., Tessarollo L., Kaldis P., Chem. J. Chinese Universities,2010, 31, 1775-1785
|
| [9] |
Malumbres M., Sotillo R. O., Santamarí A. D., Galán J., Cerezo A., Ortega S., Dubus P., Barbacid M., Cell,2004, 118, 493-504
|
| [10] |
Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J. F., Dubus P., Malumbres M., Barbacid M., Nature,2007, 448, 811-815
|
| [11] |
Shao H., Shi S., Huang S., Hole A., Abbas A. Y., Baumli S., Liu X., Lam F., Foley D., Fischer P. M., Chem. J. Chinese Universities,2010, 31, 640-659
|
| [12] |
Sansó M., Levin R. S., Lipp J. J., Wang V. Y., Greifenberg A. K., Quezada E. M., Ali A., Ghosh A., Larochelle S., Rana T. M., Chem. J. Chinese Universities,2010, 31, 117-129
|
| [13] |
Huang C. H., Lujambio A., Zuber J., Tschaharganeh D. F., Doran M. G., Evans M. J., Kitzing T., Zhu N., De S. E., Sawyers C. L., Genes Dev., 2014, 28, 1800-1813
|
| [14] |
Walsby E., Pratt G., Shao H., Abbas A. Y., Fischer P. M., Bradshaw T. D., Brennan P., Fegan C., Wang S., Pepper C., Oncotarget,2014, 5, 375-382
|
| [15] |
Mariaule G., Belmont P., Molecules,2014, 19, 14366-14382
|
| [16] |
Manohar S. M., Rathos M. J., Sonawane V., Rao S. V., Joshi K. S., Chem. J. Chinese Universities,2010, 31, 821-833
|
| [17] |
Heathcote D. A., Patel H., Kroll S. H., Hazel P., Periyasamy M., Alikian M., Kanneganti S. K., Jogalekar A. S., Scheiper B., Barbazanges M., Chem. J. Chinese Universities,2010, 31, 8508-8522
|
| [18] |
Conroy A., Stockett D. E., Walker D., Arkin M. R., Hoch U., Fox J. A., Hawtin R. E., Cancer Chemotherapy and Pharmacolog,2009, 64, 723-732
|
| [19] |
Wu Y., Chen C., Sun X., Shi X., Jin B., Ding K., Yeung S.C., Pan J., Clin. Cancer Res., 2012, 18, 1966-1978
|
| [20] |
Bettayeb K., Tirado O. M., Marionneaulambot S., Ferandin Y., Lozach O., Morris J. C., Mateolozano S., Drueckes P., Schächtele C., Kubbutat M. H., Chem. J. Chinese Universities,2010, 31, 8325-8334
|
| [21] |
Albert T. K., Rigault C., Eickhoff J., Baumgart K., Antrecht C., Klebl B., Mittler G., Meisterernst M., Chem. J. Chinese Universities,2010, 31, 55-68
|
| [22] |
Sawant R. R., Jhaveri A. M., Koshkaryev A., Lin Z., Qureshi F., Torchilin V. P., Chem. J. Chinese Universities,2010, 31, 375-388
|
| [23] |
Sonawane Y.A., Taylor M. A., Napoleon J. V., Rana S., Contreras J. I.,Journal of Medicinal Chemistry, 2016, 59-67
|
| [24] |
Kryštof V., Baumli S., Fürst R., Chem. J. Chinese Universities,2010, 31, 2883-2890
|
| [25] |
Lu H., Xue Y., Xue Y., Yu G. K., Arias C., Lin J., Fong S., Faure M., Weisburd B., Ji X., Chem. J. Chinese Universities,2010, 31
|
| [26] |
Wu S. Y., Mcnae I., Kontopidis G., Mcclue S. J., Mcinnes C., Stewart K. J., Wang S., Zheleva D. I., Marriage H., Lane D. P., Struct., 2003, 11, 399-410
|
| [27] |
Wang S., Meades C., Wood G., Osnowski A., Anderson S., Yuill R., Thomas M., Mezna M., Jackson W., Midgley C., Chem. J. Chinese Universities,2010, 31, 1662-1670
|
| [28] |
Wang S., Griffiths G., Midgley C. A., Barnett A. L., Cooper M., Grabarek J., Ingram L., Jackson W., Kontopidis G., Mcclue S. J., Chem. J. Chinese Universities,2010, 31, 1111-1121
|
| [29] |
Lukasik P. M., Elabar S., Lam F., Shao H., Liu X., Abbas A. Y., Wang S., European Journal of Medicinal Chemistry,2012, 57, 311-322
|
| [30] |
Shao H., Shi S., Foley D. W., Lam F., Abbas A. Y., Liu X., Huang S., Jiang X., Baharin N., Fischer P. M., European Journal of Medicinal Chemistry,2013, 70, 447-455
|
| [31] |
Németh G., Varga Z., Greff Z., Bencze G., Sipos A., Szántaikis C., Baska F., Gyuris A., Kelemenics K., Szathmáry Z., Chem. J. Chinese Universities,2010, 31, 342-358
|
| [32] |
Nemeth G., Greff Z., Sipos A., Varga Z., Szekely R., Sebestyen M., Jaszay Z., Beni S., Nemes Z., Pirat J. L., Volle J. N., Virieux D., Gyuris A., Kelemenics K., Ay E., Minarovits J., Szathmary S., Keri G., Orfi L., J. Med. Chem., 2014, 57, 3939-3965
|
| [33] |
Zhang Q. Q., Yao Q. Z., Zhang S. P., Bi L. M., Zhou Z. G., Zhang J., Acta Phys. Chim. Sin., 2014, 30, 371-381
|
|
(张青青, 姚其正, 张生平, 毕乐明, 周之光, 张骥. 物理化学学报, 2014, 30, 371-381)
|
| [34] |
Liu B. G., Liu J. W., Li J. Q., Geng S., Mo H. Z., Liang G. Z., Chem. J. Chinese Universities., 2017, 38(1), 41-46
|
|
(刘本国, 刘江伟, 李嘉琪, 耿升, 莫海珍, 梁桂兆. 高等学校化学学报, 2017, 38(1), 41-46)
|
| [35] |
Chen W. Y., Wang S. S., Li D. L., Peng W., Zhao J. F., Duan H. X., Chem. J. Chinese Universities., 2013, 34(12), 2798-2805
|
|
陈文雅, 王珊珊, 李冬玲, 彭炜, 赵静馥, 段红霞. 高等学校化学学报, 2013, 34(12), 2798-2805
|
| [36] |
Chen Y., Cai X., Jiang L., Li Y., Ecotoxicology and Chem. J. Chinese Universities,2010, 31, 202-215
|
| [37] |
Li B., Zhou R., He G., Guo L., Huang W., Chem. J. Chinese Universities,2010, 31, 1396-1403
|
|
(李博, 周锐, 何谷, 郭丽, 黄维. 化学学报, 2013, 71, 1396-1403)
|
| [38] |
Czudnochowski N., Bösken C. A., Geyer M., Chem. J. Chinese Universities,2010, 31, 842-854
|
| [39] |
Hole A. J., Baumli S., Shao H., Shi S., Huang S., Pepper C., Fischer P. M., Wang S., Endicott J. A., Noble M. E., Chem. J. Chinese Universities,2010, 31, 660-670
|
| [40] |
Baumli S., Hole A. J., Noble M. E., Endicott J. A., Chem. J. Chinese Universities,2010, 31, 811-822
|
| [41] |
Li X., Ye L., Wang X., Shi W., Qian X., Zhu Y., Yu H., Chem. J. Chinese Universities,2010, 31, 357-367
|
 )
)