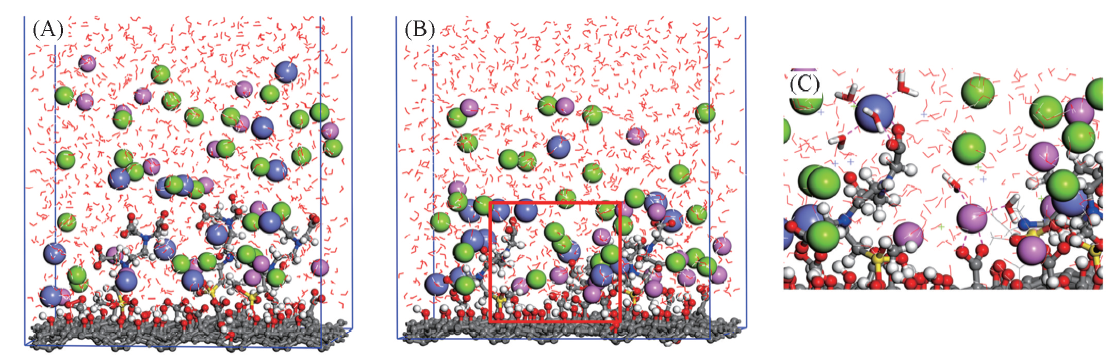
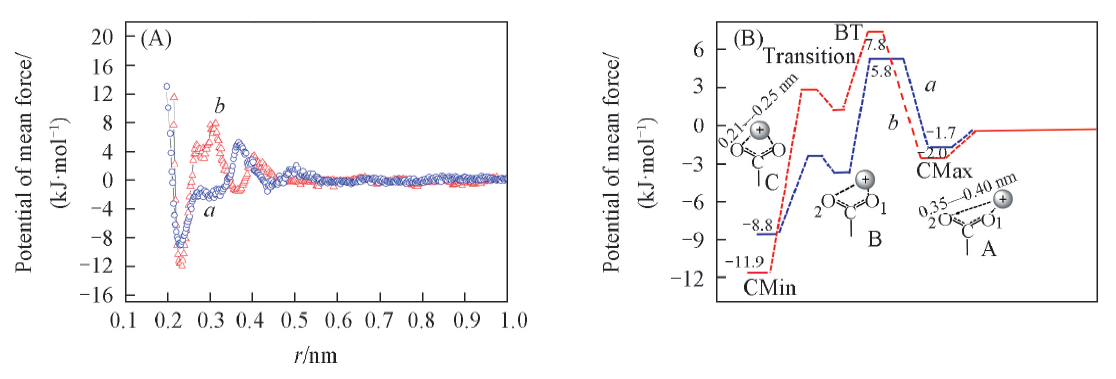
| [1] |
Agrawal C. M., Ray R. B., Journal of Biomedical Materials Research,2001, 55, 141—150
|
| [2] |
Guelcher S. A., Tissue Engineering Part B: Reviews, 2008, 14, 3—17
|
| [3] |
Lerf A., He H., Forster M., Klinowski J., The Journal of Physical Chemistry B,1998, 102, 4477—4482
|
| [4] |
Dreyer D. R., Park S., Bielawski C. W., Ruoff R. S., Chemical Society Reviews,2010, 39, 228—240
|
| [5] |
Mkhoyan K. A., Contryman A. W., Silcox J., Stewart D. A., Eda G., Mattevi C., Miller S., Chhowalla M., Nano Letters,2009, 9, 1058—1063
|
| [6] |
Zhao G., Li J., Ren X., Chen C., Wang X., Environmental Science & Technology,2011, 45, 10454—10462
|
| [7] |
Lee J. H., Kim B. S., Lee J. C., Park S., Materials Science Forum,2005, 486/487, 510—513
|
| [8] |
Repo E., Warchoł J. K., Bhatnagar A., Sillanpää M., Journal of Colloid and Interface Science,2011, 358, 261—267
|
| [9] |
Madadrang C. J., Kim H. Y., Gao G., Wang N., Zhu J., Feng H., Gorring M., Kasner M. L., Hou S., ACS Applied Materials & Interfaces,2012, 4, 1186—1193
|
| [10] |
Wan S., He F., Wu J., Wan W., Gu Y., Gao B., Journal of Hazardous Materials,2016, 314, 32—40
|
| [11] |
Anitha K., Namsani S., Singh J. K., The Journal of Physical Chemistry A,2015, 119, 8349—8358
|
| [12] |
Guo K., Zhang H., Yuan S. L., Liu C. B., Chemical Journal of Chinese Universities,2015, 36(11), 2171—2178
|
|
(郭凯, 苑世领, 刘成卜. 高等学校化学学报, 2015, 36(11), 2171—2178)
|
| [13] |
Ma Y., Zhang H., Yuan S. L., Chem. J. Chinese Universities,2015, 36(2), 386—394
|
|
(马莹, 张恒, 苑世领. 高等学校化学学报, 2015, 36(2), 386—394)
|
| [14] |
Jorgensen W. L., Maxwell D. S., Tirado-Rives J., Journal of the American Chemical Society,1996, 118, 11225—11236
|
| [15] |
Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J., Journal of Computational Chemistry,2005, 26, 1701—1718
|
| [16] |
Hess B., Kutzner C., David V. D. S., Lindahl E., Journal of Chemical Theory & Computation,2008, 4(3), 435—447
|
| [17] |
Accelrys Software Inc., Materials Studio, Release 4.4, Accelrys Software Inc.,San Diego, 2008
|
| [18] |
Anitha K., Namsani S., Singh J. K., Journal of Physical Chemistry A,2015, 119, 8349—8358
|
| [19] |
Zhang H., Wang H., Lin C. G., Wang L., Yuan S. L., Acta Chimica Sinica,2013, 71(4), 649—656
|
|
(张恒, 王华, 蔺存国, 王利, 苑世领. 化学学报, 2013, 71(4), 649—656)
|
| [20] |
Andre M. K., Contryman A. W., Silcox J., Stewart D. A., Eda G., Mattevi C., Miller S., Chhowalla M., Nano Letters,2009, 9, 1058—1063
|
| [21] |
Allen T. W., Andersen O. S., Roux B., Biophysical Chemistry,2006, 124, 251—267
|
| [22] |
Andre M. K., Contryman A. W., Silcox J., Stewart D. A., Eda G., Mattevi C., Miller S., Chhowalla M., Nano Letters,2009, 9, 1058—1063
|
| [23] |
Berendsen H., Grigera J., Straatsma T., Journal of Physical Chemistry,1987, 91, 6269—6271
|
 ), 侯士峰2(
), 侯士峰2(