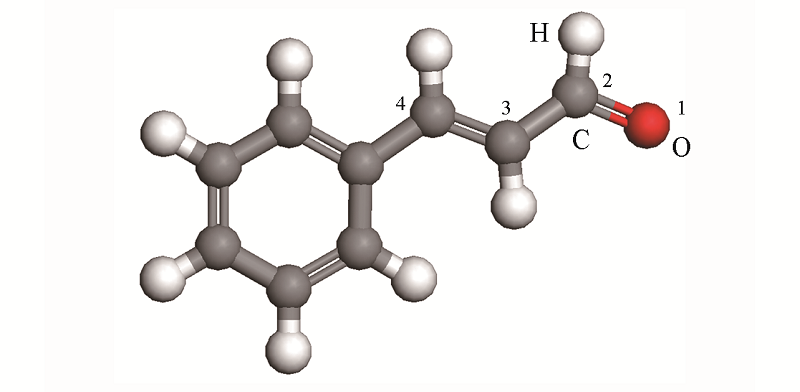
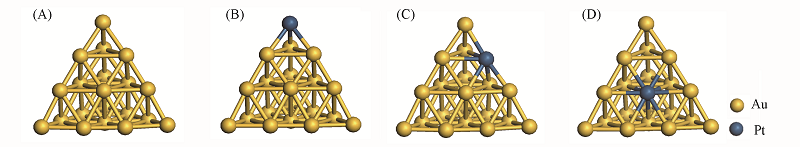
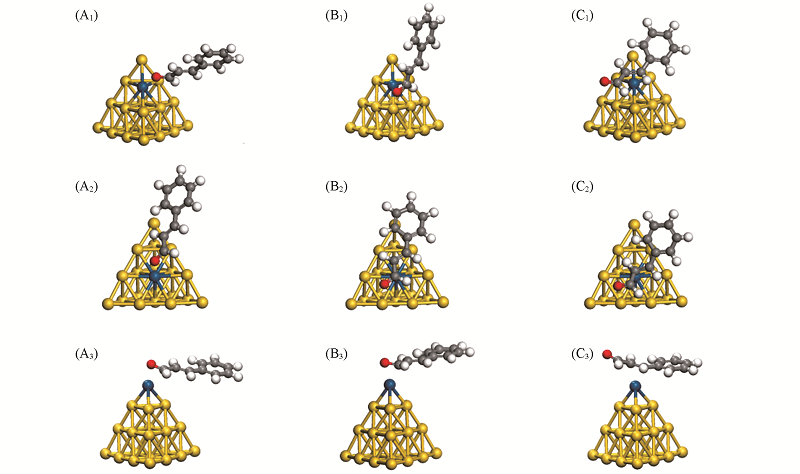
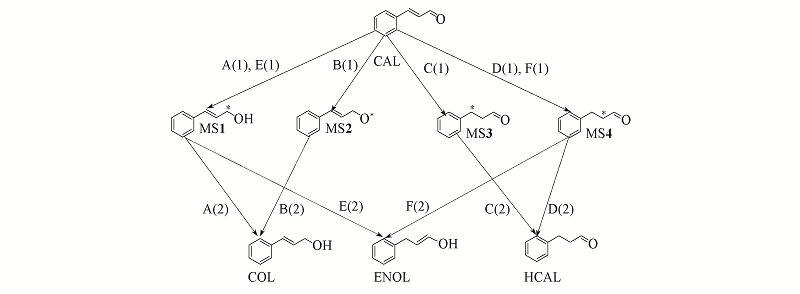
| [1] |
Lee J. D., Christopher M. A., Parlett1 N. S., Scientific Reports,2015, 5(9426), 1—7
|
| [2] |
Kolodziej M., Drelinkiewicz A., Lalik E., Gurgul J., Duraczynska D., Kosydar R., Applied Catalysis A: General,2016, 515, 60—71
|
| [3] |
Karl R., Kahsar D. K., Schwartz J., Will M., J. Am. Chem. Soc., 2014, 136, 520—526
|
| [4] |
Fang C., Chen Y. J., Mao B., Zhao J., Jiang Y. F., Zhao S. L., Ma J., Chem. J. Chinese Universities,2015, 36(1), 124—130(方超, 陈亚君, 毛卉, 赵俊, 蒋云福, 赵仕林, 马骏. 高等学校化学学报, 2015, 36(1), 124—130)
|
| [5] |
Li H., Ma C.J., Li H. X., Acta Chimica Sinica, 2006, 24, 1947—1953(李辉, 马春景, 李和兴. 化学学报, 2006, 24, 1947—1953)
|
| [6] |
Xue Q. Q., Karine P., Pierre L., Philippe S., Dalton Trans., 2014, 43, 9283—9295
|
| [7] |
Haeck J.D., Veldeman N., J. Phys.Chem. A, 2011, 115, 2103—2109
|
| [8] |
Qian H., Jiang D., Gayathri C., J. Am. Chem. Soc., 2012, 134, 16159—16162
|
| [9] |
Cheng L., Xiao Y. K., Zhi W. L., J. Phys. Chem. A,2011, 115, 9273—9281
|
| [10] |
Zhang X. D., Gao M. L., Wu D., Int. J. Mol. Sci., 2011, 12, 2972—2981
|
| [11] |
Yuan D. W., Wang Y., Zenga Z., J. Chem. Phys., 2005, 122, 114310
|
| [12] |
Mondal K., Ghanty T. K., Banerjee A., Mol. Phys., 2013, 111, 725—734
|
| [13] |
Sinfelt J. H., Accounts Chem. Res., 1987, 20, 134—136
|
| [14] |
Gholizadeh R., Yu Y. X., Appl. Surf. Sci., 2015, 357, 1187—1195
|
| [15] |
Liu T. T., Lu X., Zhang M. T., Chem. Res. Chinese Universities,2014, 30(4), 656—660
|
| [16] |
Pan W., Ma G. W., Yang X. D., Chem. J. Chinese Universities,2015, 36(2), 325—329(潘威, 马广文, 杨小东. 高等学校化学学报, 2015, 36(2), 325—329)
|
| [17] |
Tapan K. G., J. Phys. Chem. C, 2014, 118, 11935—11945
|
| [18] |
Anna V., Beletskaya D. P., Alexander F. S., J. Phys. Chem. A,2013, 117, 6817—6826
|
| [19] |
Ke Q. S., Yong C. H., Gui R. Z., ACS Catal., 2011, 1, 1336—1346
|
| [20] |
Yong C. H., Ke Q. S., Gui R. Z., Chem. Commun., 2011, 47, 1300—1302
|
| [21] |
Delley B., J. Phys. Chem. A, 2006, 110, 13632—13639
|
| [22] |
Chen W. K., Cao M. J., Liu S. H., Acta Phys-Chim Sinica,2005, 21, 903—908(陈文凯, 曹梅娟, 刘书红. 物理化学学报, 2005,21, 903—908)
|
| [23] |
Xiao X. C., Shi W., Ni Z. M., Zhang L. Y., Acta Phys-Chim Sinica,2015, 31, 885—892(肖雪春, 施炜, 倪哲明, 张连阳. 物理化学学报, 2015,31, 885—892)
|
| [24] |
Tao J., Yao Z.J., Xue F., Fundamentals of Material Science, Chemical Industry Press, 2006, 50—51
|
|
(陶杰, 姚正军, 薛烽. 化学工业出版社, 2006, 50—51)
|
| [25] |
Ham H. C., Hwang G. S., Han J., Nam S. W., Lim T. H., J. Phys. Chem. C,2010, 114, 14922—14928
|
| [26] |
Morrow B. H., Resasco D. E., Striolo A., Nardelli M. B., J. Phys. Chem. C,2011, 115, 5637—5647
|
| [27] |
Diego C. A., Maria P. O., Luis S., J. Phys. Chem. A,2015, 119, 6909—6918
|
| [28] |
Delbecq D., Sautet P., J. Catal., 1995, 152, 217—236
|
| [29] |
Mulliken R. S., J. Chem. Phys., 1955, 23, 1833—1840
|
| [30] |
Loffreda D., Delbecq F., Vigne F., Sautet P., J. Am. Chem. Soc., 2006, 128, 1316—1323
|
| [31] |
Shi W., Zhang L. Y., Ni Z. M., Xiao X. C., Xia S. J., RSC Advances,2014, 4, 27003—27012
|
| [32] |
Zhang L.Y., Jiang J. H., Shi W., Xia S. J., Ni Z. M., RSC Advances,2015, 5, 34319—34326
|
| [33] |
Jiang J. H., Xia S. J., Ni Z. M., Zhang L.Y., Chem. J. Chinese Universities,2016, 37(4), 693—700(蒋军辉, 夏盛杰, 倪哲明, 张连阳. 高等学校化学学报, 2016, 37(4), 693—700)
|
 ), 夏盛杰, 钱梦丹, 薛继龙
), 夏盛杰, 钱梦丹, 薛继龙