

高等学校化学学报 ›› 2016, Vol. 37 ›› Issue (8): 1468.doi: 10.7503/cjcu20160307
于泳博, 刘翠, 宫利东( )
)
收稿日期:2016-05-03
出版日期:2016-07-19
发布日期:2016-07-19
作者简介:联系人简介: 宫利东, 男, 博士, 教授, 主要从事理论与计算化学研究. E-mail:
基金资助:
YU Yongbo, LIU Cui, GONG Lidong*()
Received:2016-05-03
Online:2016-07-19
Published:2016-07-19
Contact:
GONG Lidong
E-mail:gongjw@lnnu.edu.cn
Supported by:摘要:
采用从头算方法(ab initio)和原子-键电负性均衡浮动电荷分子力场方法(ABEEMσπ/MM), 对甲醇团簇(CH3OH)n(n=3~12)和[Na(CH3OH)n]+(n=3~6)体系的结构、 电荷分布和结合能进行研究. 依据从头算结果构建上述体系的ABEEMσπ/MM浮动电荷势能函数, 并确定相关参数. 结果表明, ABEEMσπ/MM所获得的结构和结合能等均优于OPLS/AA力场, 并与从头算结果相符, 其中键长的平均绝对偏差(AAD)小于0.004 nm, 键长、 键角和结合能的相对均方根偏差(RRMSD)分别小于3.8%, 1.7%和6.8%; 电荷分布与从头算结果的线性相关系数均大于0.99.
中图分类号:
TrendMD:
于泳博, 刘翠, 宫利东. 从头算和ABEEMσπ/MM对(CH3OH)n(n=3~12)和[Na(CH3OH)n]+(n=3~6)体系的研究. 高等学校化学学报, 2016, 37(8): 1468.
YU Yongbo,LIU Cui,GONG Lidong. Studies of (CH3OH)n(n=3—12) and [Na(CH3OH)n]+(n=3—6)via ab initio and ABEEMσπ/MM†. Chem. J. Chinese Universities, 2016, 37(8): 1468.
| Code | χ* | 2η* | σ | ε |
|---|---|---|---|---|
| C | 3.150 | 4.700 | 3.500 | 0.066 |
| H(—H3C) | 2.455 | 13.038 | 2.500 | 0.030 |
| O | 3.300 | 4.327 | 3.120 | 0.170 |
| H(—HO) | 2.060 | 6.300 | 1.450 | 0.010 |
| σ(C—H) | 6.010 | 42.350 | ||
| σ(C—O) | 6.920 | 49.780 | ||
| σ(O—H) | 6.050 | 62.180 | ||
| Olp | 4.035 | 5.730 | ||
| Na+ | 8.101 | 14.510 | 3.330 | 0.003 |
Table 1 ABEEMσπ/MM parameters(χ*, 2η*, σ, ε) of (CH3OH)n and Na+(CH3OH)n
| Code | χ* | 2η* | σ | ε |
|---|---|---|---|---|
| C | 3.150 | 4.700 | 3.500 | 0.066 |
| H(—H3C) | 2.455 | 13.038 | 2.500 | 0.030 |
| O | 3.300 | 4.327 | 3.120 | 0.170 |
| H(—HO) | 2.060 | 6.300 | 1.450 | 0.010 |
| σ(C—H) | 6.010 | 42.350 | ||
| σ(C—O) | 6.920 | 49.780 | ||
| σ(O—H) | 6.050 | 62.180 | ||
| Olp | 4.035 | 5.730 | ||
| Na+ | 8.101 | 14.510 | 3.330 | 0.003 |
| Interaction site | A | B | C | D |
|---|---|---|---|---|
| lp of O in methanol and H | 0.5509 | 0.0462 | 1.12963 | 0.01638 |
| lp of O in methanol and Na+ | 0.8660 | 0.2220 | 1.83031 | 0.02248 |
Table 2 Parameters of klp,H
| Interaction site | A | B | C | D |
|---|---|---|---|---|
| lp of O in methanol and H | 0.5509 | 0.0462 | 1.12963 | 0.01638 |
| lp of O in methanol and Na+ | 0.8660 | 0.2220 | 1.83031 | 0.02248 |
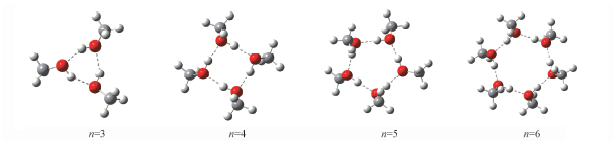
Fig.1 Stable geometry of cyclic (CH3OH)n(n=3—6) optimized via ab initio and ABEEMσπ/MM
| n | ROH/nm | RO…H/nm | ||||||
|---|---|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | |
| 3 | 0.0978 | 0.0947 | 0.0967 | 0.0984 | 0.1883 | 0.1897 | 0.1838 | |
| 4 | 0.0986 | 0.0950 | 0.0972 | 0.0991 | 0.1740 | 0.1772 | 0.1759 | |
| 5 | 0.0988 | 0.0950 | 0.0973 | 0.0991 | 0.1706 | 0.1741 | 0.1758 | |
| 6 | 0.0989 | 0.0948 | 0.0973 | 0.0990 | 0.1694 | 0.1731 | 0.1780 | |
| AAD | 0.0036 | 0.0014 | 0.0004 | 0.0030 | 0.0051 | |||
| RRMSD | 3.72% | 1.43% | 0.43% | 1.76% | 3.18% | |||
| n | RO…O/nm | ∠O1—H1—O2/(°) | ||||||
| QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | |
| 3 | 0.2775 | 0.2812 | 0.2747 | 0.2799 | 150.24 | 150.93 | 155.37 | 153.00 |
| 4 | 0.2712 | 0.2709 | 0.2719 | 0.2754 | 168.12 | 168.11 | 168.56 | 168.70 |
| 5 | 0.2692 | 0.2690 | 0.2728 | 0.2752 | 175.65 | 175.62 | 174.45 | 174.50 |
| 6 | 0.2681 | 0.2679 | 0.2751 | 0.2752 | 176.71 | 176.63 | 176.63 | 176.70 |
| AAD | 0.0011 | 0.0035 | 0.0049 | 0.20 | 1.71 | 1.13 | ||
| RRMSD | 0.68% | 1.54% | 1.93% | 0.21% | 1.57% | 0.91% | ||
Table 3 ROH, RO…H, RO…O and ∠O1—H1—O2 of cyclic (CH3OH)n(n=3—6)
| n | ROH/nm | RO…H/nm | ||||||
|---|---|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | |
| 3 | 0.0978 | 0.0947 | 0.0967 | 0.0984 | 0.1883 | 0.1897 | 0.1838 | |
| 4 | 0.0986 | 0.0950 | 0.0972 | 0.0991 | 0.1740 | 0.1772 | 0.1759 | |
| 5 | 0.0988 | 0.0950 | 0.0973 | 0.0991 | 0.1706 | 0.1741 | 0.1758 | |
| 6 | 0.0989 | 0.0948 | 0.0973 | 0.0990 | 0.1694 | 0.1731 | 0.1780 | |
| AAD | 0.0036 | 0.0014 | 0.0004 | 0.0030 | 0.0051 | |||
| RRMSD | 3.72% | 1.43% | 0.43% | 1.76% | 3.18% | |||
| n | RO…O/nm | ∠O1—H1—O2/(°) | ||||||
| QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | QMa | ABEEMσπ/MM | OPLS/AAb | ONIOMc | |
| 3 | 0.2775 | 0.2812 | 0.2747 | 0.2799 | 150.24 | 150.93 | 155.37 | 153.00 |
| 4 | 0.2712 | 0.2709 | 0.2719 | 0.2754 | 168.12 | 168.11 | 168.56 | 168.70 |
| 5 | 0.2692 | 0.2690 | 0.2728 | 0.2752 | 175.65 | 175.62 | 174.45 | 174.50 |
| 6 | 0.2681 | 0.2679 | 0.2751 | 0.2752 | 176.71 | 176.63 | 176.63 | 176.70 |
| AAD | 0.0011 | 0.0035 | 0.0049 | 0.20 | 1.71 | 1.13 | ||
| RRMSD | 0.68% | 1.54% | 1.93% | 0.21% | 1.57% | 0.91% | ||
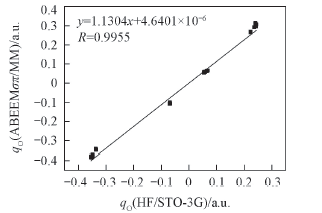
Fig.2 Charge linear correlation diagram of (CH3OH)n(n=3—6) via ab initio and ABEEMσπ/MM
| n | Δ | |||||
|---|---|---|---|---|---|---|
| QMb | ABEEMσπ/MM | OPLS/AAc | Cluster approach ad | Cluster approach bd | PHH3e | |
| 3 | 61.24 | 63.41 | 55.05 | 64.83 | 66.92 | 59.11 |
| 4 | 112.11 | 115.41 | 89.70 | 124.98 | 131.88 | 98.27 |
| 5 | 151.57 | 169.50 | 146.72 | 174.52 | 186.51 | 132.30 |
| 6 | 188.85 | 189.35 | 215.77 | 221.08 | 237.76 | |
| AAD | 5.97 | 15.09 | 17.91 | 27.32 | ||
| RRMSD | 6.71% | 13.11% | 15.25% | 23.20% | ||
Table 4 Binding energy(ΔEn) of cyclic methanol cluster(CH3OH)n(n=3—6) applied by MP2, ABEEMσπ/MM, OPLS/AA, PHH3 and cluster approaches
| n | Δ | |||||
|---|---|---|---|---|---|---|
| QMb | ABEEMσπ/MM | OPLS/AAc | Cluster approach ad | Cluster approach bd | PHH3e | |
| 3 | 61.24 | 63.41 | 55.05 | 64.83 | 66.92 | 59.11 |
| 4 | 112.11 | 115.41 | 89.70 | 124.98 | 131.88 | 98.27 |
| 5 | 151.57 | 169.50 | 146.72 | 174.52 | 186.51 | 132.30 |
| 6 | 188.85 | 189.35 | 215.77 | 221.08 | 237.76 | |
| AAD | 5.97 | 15.09 | 17.91 | 27.32 | ||
| RRMSD | 6.71% | 13.11% | 15.25% | 23.20% | ||
| n | ROH/nm | RO…H/nm | ||||
|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 7 | 0.0953 | 0.0951 | 0.0972 | 0.1881 | 0.1874 | 0.1803 |
| 8 | 0.0953 | 0.0951 | 0.0971 | 0.1877 | 0.1867 | 0.1830 |
| 9 | 0.0953 | 0.0951 | 0.0971 | 0.1879 | 0.1868 | 0.1840 |
| 10 | 0.0953 | 0.0951 | 0.0970 | 0.1882 | 0.1873 | 0.1866 |
| 11 | 0.0953 | 0.0946 | 0.0970 | 0.1878 | 0.1878 | 0.1882 |
| 12 | 0.0953 | 0.0947 | 0.0966 | 0.1881 | 0.1888 | |
| AAD | 0.0004 | 0.0017 | 0.0007 | 0.0037 | ||
| RRMSD | 0.43% | 1.80% | 0.43% | 2.39% | ||
| n | RO…O/nm | ∠O1—H1—O2/(°) | ||||
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 7 | 0.2832 | 0.2824 | 0.2773 | 175.54 | 178.39 | 176.43 |
| 8 | 0.2828 | 0.2817 | 0.2799 | 174.55 | 177.89 | 175.08 |
| 9 | 0.2830 | 0.2818 | 0.2809 | 174.55 | 177.69 | 175.11 |
| 10 | 0.2832 | 0.2823 | 0.2834 | 175.07 | 177.45 | 174.95 |
| 11 | 0.2829 | 0.2824 | 0.2849 | 175.14 | 178.05 | 174.24 |
| 12 | 0.2831 | 0.2834 | 175.24 | 177.51 | ||
| AAD | 0.0008 | 0.0026 | 2.81 | 0.6 | ||
| RRMSD | 0.30% | 1.14% | 1.62% | 0.38% | ||
Table 5 ROH, RO…H, RO…O and ∠O1—H1—O2 of cyclic (CH3OH)n(n=7—12)
| n | ROH/nm | RO…H/nm | ||||
|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 7 | 0.0953 | 0.0951 | 0.0972 | 0.1881 | 0.1874 | 0.1803 |
| 8 | 0.0953 | 0.0951 | 0.0971 | 0.1877 | 0.1867 | 0.1830 |
| 9 | 0.0953 | 0.0951 | 0.0971 | 0.1879 | 0.1868 | 0.1840 |
| 10 | 0.0953 | 0.0951 | 0.0970 | 0.1882 | 0.1873 | 0.1866 |
| 11 | 0.0953 | 0.0946 | 0.0970 | 0.1878 | 0.1878 | 0.1882 |
| 12 | 0.0953 | 0.0947 | 0.0966 | 0.1881 | 0.1888 | |
| AAD | 0.0004 | 0.0017 | 0.0007 | 0.0037 | ||
| RRMSD | 0.43% | 1.80% | 0.43% | 2.39% | ||
| n | RO…O/nm | ∠O1—H1—O2/(°) | ||||
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 7 | 0.2832 | 0.2824 | 0.2773 | 175.54 | 178.39 | 176.43 |
| 8 | 0.2828 | 0.2817 | 0.2799 | 174.55 | 177.89 | 175.08 |
| 9 | 0.2830 | 0.2818 | 0.2809 | 174.55 | 177.69 | 175.11 |
| 10 | 0.2832 | 0.2823 | 0.2834 | 175.07 | 177.45 | 174.95 |
| 11 | 0.2829 | 0.2824 | 0.2849 | 175.14 | 178.05 | 174.24 |
| 12 | 0.2831 | 0.2834 | 175.24 | 177.51 | ||
| AAD | 0.0008 | 0.0026 | 2.81 | 0.6 | ||
| RRMSD | 0.30% | 1.14% | 1.62% | 0.38% | ||
| n | Δ | n | Δ | ||||
|---|---|---|---|---|---|---|---|
| QMb | ABEEMσπ/MM | OPLS/AAc | QMb | ABEEMσπ/MM | OPLS/AAc | ||
| 7 | 220.04 | 222.92 | 295.11 | 11 | 353.63 | 328.97 | 672.65 |
| 8 | 252.85 | 241.31 | 378.54 | 12 | 390.16 | 373.98 | |
| 9 | 283.95 | 266.22 | 473.34 | AAD | 14.26 | 192.16 | |
| 10 | 323.20 | 310.62 | 574.83 | RRMSD | 5.08% | 72.55% | |
Table 6 ΔEn of cyclic (CH3OH)n(n=7—12)
| n | Δ | n | Δ | ||||
|---|---|---|---|---|---|---|---|
| QMb | ABEEMσπ/MM | OPLS/AAc | QMb | ABEEMσπ/MM | OPLS/AAc | ||
| 7 | 220.04 | 222.92 | 295.11 | 11 | 353.63 | 328.97 | 672.65 |
| 8 | 252.85 | 241.31 | 378.54 | 12 | 390.16 | 373.98 | |
| 9 | 283.95 | 266.22 | 473.34 | AAD | 14.26 | 192.16 | |
| 10 | 323.20 | 310.62 | 574.83 | RRMSD | 5.08% | 72.55% | |
| n | y=Ax+Ba | R | S | Umax | qO/a.u. | |
|---|---|---|---|---|---|---|
| ab initio | ABEEM | |||||
| 7 | y=1.0923x-2.7971×10-6 | 0.9933 | 0.0233 | 0.0431 | -0.3590 | -0.3675 |
| 8 | y=1.0867x-8.7182×10-6 | 0.9937 | 0.0226 | 0.0386 | -0.3595 | -0.3666 |
| 9 | y=1.0829x-1.3304×10-6 | 0.9936 | 0.0225 | 0.0434 | -0.3594 | -0.3654 |
| 10 | y=1.0837x-1.7028×10-6 | 0.9939 | 0.0222 | 0.0422 | -0.3593 | -0.3664 |
| 11 | y=1.0791x-6.1424×10-6 | 0.9940 | 0.0219 | 0.0445 | -0.3596 | -0.3653 |
| 12 | y=1.0796x-1.4995×10-8 | 0.9936 | 0.0225 | 0.0441 | -0.3593 | -0.3645 |
| AAD | 0.0066 | |||||
Table 7 Comparison of charge distributions for cyclic (CH3OH)n(n=7—12)*
| n | y=Ax+Ba | R | S | Umax | qO/a.u. | |
|---|---|---|---|---|---|---|
| ab initio | ABEEM | |||||
| 7 | y=1.0923x-2.7971×10-6 | 0.9933 | 0.0233 | 0.0431 | -0.3590 | -0.3675 |
| 8 | y=1.0867x-8.7182×10-6 | 0.9937 | 0.0226 | 0.0386 | -0.3595 | -0.3666 |
| 9 | y=1.0829x-1.3304×10-6 | 0.9936 | 0.0225 | 0.0434 | -0.3594 | -0.3654 |
| 10 | y=1.0837x-1.7028×10-6 | 0.9939 | 0.0222 | 0.0422 | -0.3593 | -0.3664 |
| 11 | y=1.0791x-6.1424×10-6 | 0.9940 | 0.0219 | 0.0445 | -0.3596 | -0.3653 |
| 12 | y=1.0796x-1.4995×10-8 | 0.9936 | 0.0225 | 0.0441 | -0.3593 | -0.3645 |
| AAD | 0.0066 | |||||
| n | RM…O/nm | ∠M—O1—C1/(°) | ||||
|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 3 | 0.2310 | 0.2334 | 0.2365 | 128.43 | 127.19 | 100.17 |
| 4 | 0.2346 | 0.2374 | 0.2398 | 125.33 | 123.65 | 100.65 |
| 5 | 0.2342 | 0.2377 | 0.2397 | 129.03 | 126.66 | 100.37 |
| 6 | 0.2338 | 0.2346 | 0.2402 | 134.96 | 134.34 | 106.25 |
| AAD | 0.0024 | 0.0057 | 1.48 | 27.58 | ||
| RRMSD | 1.10% | 2.43% | 1.24% | 21.34% | ||
| n | ∠O1—M—O2/(°) | Δ | ||||
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 3 | 119.87 | 119.88 | 119.99 | 250.59 | 226.68 | 520.79 |
| 4 | 104.82 | 105.00 | 109.47 | 309.99 | 297.16 | 602.51 |
| 5 | 98.97 | 98.81 | 110.66 | 367.76 | 378.08 | 651.70 |
| 6 | 93.81 | 93.79 | 110.64 | 423.10 | 457.84 | 686.82 |
| AAD | 0.09 | 8.32 | 20.45 | 277.60 | ||
| RRMSD | 0.12% | 10.02% | 6.58% | 80.78% | ||
Table 8 RM…O, ∠M—O1—C1, ∠O1—M—O2 and ΔEn of [Na(CH3OH)n]+(n=3—6)
| n | RM…O/nm | ∠M—O1—C1/(°) | ||||
|---|---|---|---|---|---|---|
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 3 | 0.2310 | 0.2334 | 0.2365 | 128.43 | 127.19 | 100.17 |
| 4 | 0.2346 | 0.2374 | 0.2398 | 125.33 | 123.65 | 100.65 |
| 5 | 0.2342 | 0.2377 | 0.2397 | 129.03 | 126.66 | 100.37 |
| 6 | 0.2338 | 0.2346 | 0.2402 | 134.96 | 134.34 | 106.25 |
| AAD | 0.0024 | 0.0057 | 1.48 | 27.58 | ||
| RRMSD | 1.10% | 2.43% | 1.24% | 21.34% | ||
| n | ∠O1—M—O2/(°) | Δ | ||||
| QMa | ABEEMσπ/MM | OPLS/AAb | QMa | ABEEMσπ/MM | OPLS/AAb | |
| 3 | 119.87 | 119.88 | 119.99 | 250.59 | 226.68 | 520.79 |
| 4 | 104.82 | 105.00 | 109.47 | 309.99 | 297.16 | 602.51 |
| 5 | 98.97 | 98.81 | 110.66 | 367.76 | 378.08 | 651.70 |
| 6 | 93.81 | 93.79 | 110.64 | 423.10 | 457.84 | 686.82 |
| AAD | 0.09 | 8.32 | 20.45 | 277.60 | ||
| RRMSD | 0.12% | 10.02% | 6.58% | 80.78% | ||
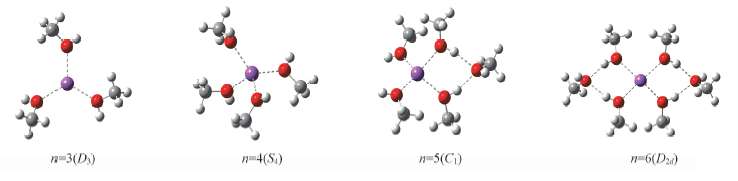
Fig.3 Stable geometry of [Na(CH3OH)n]+(n=3—6) optimized via ab initio and ABEEMσπ/MM
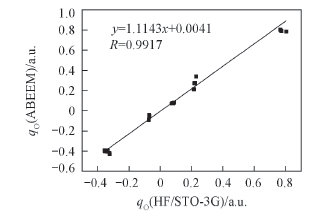
Fig.4 Charge linear correlation diagram of [Na(CH3OH)n]+(n=3—6) via ab initio and ABEEMσπ/MM
| [1] | Mountain R D., Int. J Thermophys., 2007, 28, 536—543 |
| [2] | Yizhak M., Chem Rev., 2009, 109, 1346—1370 |
| [3] | Jun S., Isao Y., Koji A., Langmuir,2010, 26, 8030—8035 |
| [4] | Medhekar N V., Ramasubramaniam A., Ruoff R S., Shenoy V B., ACS Nano,2010, 4, 2300—2306 |
| [5] | Marcus Y., Pure Appl Chem., 2010, 82, 1889—1899 |
| [6] | Boyd S L., Boyd R J., J. Chem. Theory Comput., 2007, 3, 54—61 |
| [7] | Pires M M., de Turi V F., J. Chem. Theory Comput., 2007, 3, 1073—1082 |
| [8] | Reddy A S., Sastry G N., J. Phys. Chem A,2005, 109, 8893—8903 |
| [9] | Rai D., Kulkarni A D., Gejji S P., Pathak R K., J. Chem Phys., 2011, 135, 024307 |
| [10] |
Ponder J W., Case D A., Adv. Protein Chem., 2003, 66, 27—85
doi: 10.1016/S0092-8674(03)00035-7 URL pmid: 12553913 |
| [11] |
Hart K., Foloppe N., Baker C M., Denning E J., Nilsson L., MacKerell A D., New Phytologist,2012, 8, 348—362
doi: 10.3969/j.issn.1005-2860.2012.12.002 URL |
| [12] | Christen M., Hunenberger P H., Bakowies D., Baron R., Bürgi R., Geerke D P., Heinz T N., Kastenholz M A., Kräutler V., Oostenbrink C., Peter C., Trzesniak D., Gunsteren W. F V., J. Comput Chem., 2005, 26, 1719—1751 |
| [13] | Kaminski G A., Friesner R A., J. Phys. Chem B,2001, 105, 6474—6487 |
| [14] | Do H., Besley N A., J. Chem Phys., 2012, 137, 134106 |
| [15] | Huang Z G., Yu L., Dai Y M., Struct Chem., 2010, 21, 565—572 |
| [16] | Kazachenko S., Bulusu S., Thakkar A J., J. Chem Phys., 2013, 138, 224303 |
| [17] | Morrone J A., Tuckerman M E., Chem. Phys Lett., 2003, 370, 406—411 |
| [18] | Zeng Y P., Hu J M., Yuan Y., Zhang X B., Ju S G., Chem. Phys Lett., 2012, 538, 60—66 |
| [19] | Jorgensen W L., Bigot B., Chandrasekhar J., J. Am. Chem Soc., 1982, 104, 4584—4591 |
| [20] | Forck R M., Dauster I., Buck U., Zeuch T., J. Phys. Chem A,2011, 115, 6068—6076 |
| [21] | Chen A A., Pappu R V., J. Phys. Chem B,2007, 111, 11884—11887 |
| [22] | Yang Z Z., Wang C S., J. Phys. Chem A,1997, 101, 6315—6321 |
| [23] | Yang Z Z., Cui B Q., J. Chem. Theory Comput., 2007, 3, 1561—1568 |
| [24] | Yang Z Z., Wu Y., Zhao D X., J. Chem Phys., 2004, 120, 2541—2557 |
| [25] | Zhao D X., Liu C., Wang F F., Yu C Y., Gong L D., Liu S B., Yang Z Z., J. Chem. Theory Comput., 2010, 6, 795—804 |
| [26] | Gong L D., Sci. China Chem., 2012, 55(12), 2471—2484 |
| [27] | Lu L N., Liu C., Gong L D., Chem. Res. Chinese Universities,2013, 29(2), 344—350 |
| [28] | Liu L L., Yang Z Z., Chem. J. Chinese Universities,2015, 36(11), 2179—2188 |
| [29] | Gong L D., Ren W H., Zhang Y L., Li W B., Yang Z Z., Sci. Sinica Chim., 2016, 46, 114—125 |
| [30] | Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., et al., Gaussian 09, D.01, Gaussian Inc., Wallingford CT, 2013 |
| [31] | Dennington R., Keith T., Millam J., GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 |
| [32] | Meher B R., Kumar M. V S., Bandyopadhyay P., Indian J Phys., 2009, 83, 81—90 |
| [33] | Yang Z Z., Cui B Q., J. Chem. Theory Comput., 2007, 3, 1561—1568 |
| [34] | Zhao D X., Liu C., Wang F F., Yu C Y., Gong L D., Liu S B., Yang Z Z., J. Chem. Theory Comput., 2010, 6, 795—804 |
| [35] | Yang Z Z., Wang J J., Zhao D X., J. Comput Chem., 2014, 35, 1690—1706 |
| [36] | Haynes W.M., Lide D. R., CRC Handbook of Chemistry and Physics, 95th Edition, Boca Raton, CRC Press, 2014, 9—40 |
| [37] | Buck U., Siebers J G., J. Chem Phys., 1998, 108, 20—32 |
| [38] | Hagemeister F C., Gruenloh C J., Zwier T S., J. Phys. Chem A,1998, 102, 82—94 |
| [1] | 周紫璇, 杨海艳, 孙予罕, 高鹏. 二氧化碳加氢制甲醇多相催化剂研究进展[J]. 高等学校化学学报, 2022, 43(7): 20220235. |
| [2] | 刘杰, 李金晟, 柏景森, 金钊, 葛君杰, 刘长鹏, 邢巍. 降低直接甲醇燃料电池浓差极化的含磺化碳管阻水夹层的构建[J]. 高等学校化学学报, 2022, 43(11): 20220420. |
| [3] | 张伶育, 张继龙, 曲泽星. RDX分子内振动能量重分配的动力学研究[J]. 高等学校化学学报, 2022, 43(10): 20220393. |
| [4] | 徐晓龙, 方礼宁, 刘长宇, 刘敏超, 郏建波. Z型 g-C3N4/Pt/TiO2纳米管阵列复合电极的制备及光电氧化甲醇性能[J]. 高等学校化学学报, 2021, 42(9): 2926. |
| [5] | 马卓远, 汪大洋. 分子自组装单层膜的表面浸润性研究现状和展望[J]. 高等学校化学学报, 2021, 42(4): 1031. |
| [6] | 李健, 于明明, 孙源, 冯文华, 冯兆池, 吴剑峰. 水溶液pH对甲烷低温氧化制备甲醇的影响[J]. 高等学校化学学报, 2021, 42(3): 776. |
| [7] | 孙全虎, 卢天天, 何建江, 黄长水. 含异原子石墨炔基电极材料的研究进展[J]. 高等学校化学学报, 2021, 42(2): 366. |
| [8] | 刘志刚, 李家宝, 杨剑, 马浩, 王赪胤, 郭鑫, 汪国秀. 新型石墨化氮化碳/锡/氮掺杂碳复合物的制备及储钠性能[J]. 高等学校化学学报, 2021, 42(2): 633. |
| [9] | 王蔓, 王鑫, 周静, 高国华. 聚离子液体催化碳酸乙烯酯与甲醇的酯交换反应[J]. 高等学校化学学报, 2021, 42(12): 3701. |
| [10] | 郭淑佳, 王森, 张莉, 秦张峰, 王鹏飞, 董梅, 王建国, 樊卫斌. ZSM-5分子筛酸位分布及其甲醇制烯烃催化性能的定向管理与调控[J]. 高等学校化学学报, 2021, 42(1): 227. |
| [11] | 孙维鑫, 刘佳, 王家正, 张益妙, 金磊, 周剑章, 杨防祖, 吴德印, 田中群. Au/Pt纳米柱阵列电极的制备及光电催化氧化甲醇性能[J]. 高等学校化学学报, 2020, 41(12): 2788. |
| [12] | 王艳燕, 刘会贞, 韩布兴. 多相催化剂催化二氧化碳加氢合成甲醇的研究进展[J]. 高等学校化学学报, 2020, 41(11): 2393. |
| [13] | 陈淡宜, 张福梅, 何丹, 张紫媚, 钟芬, 文思妙妙, 刘祈星, 周海峰. 钌催化的手性苯基/苯并噻唑甲醇的转移氢化合成[J]. 高等学校化学学报, 2020, 41(10): 2264. |
| [14] | 尹娇, 张国强, 阎立飞, 贾东森, 郑华艳, 李忠. 反应过程中Cu物种演变对其催化甲醇氧化羰基化反应活性的影响[J]. 高等学校化学学报, 2019, 40(7): 1510. |
| [15] | 黄锐, 姚志龙, 孙培永, 张胜红. CuO-WO3-ZrO2的结构和性质对苯甲醛加氢反应催化性能的影响[J]. 高等学校化学学报, 2019, 40(5): 1005. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||