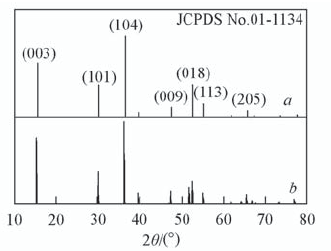
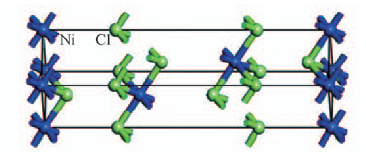
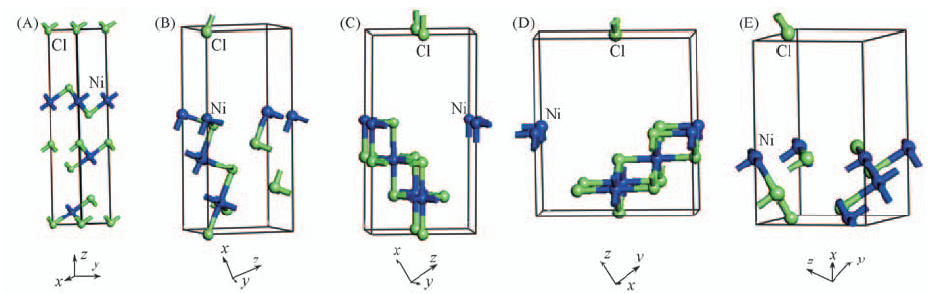
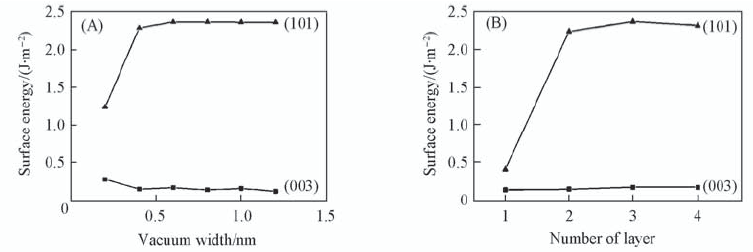
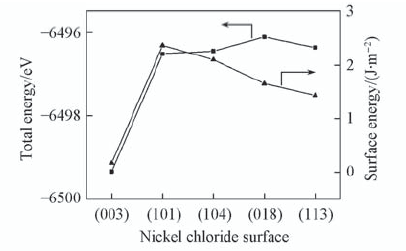
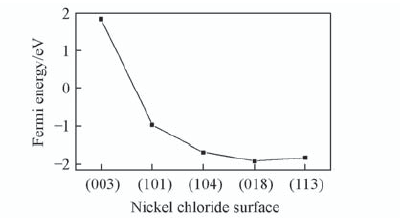
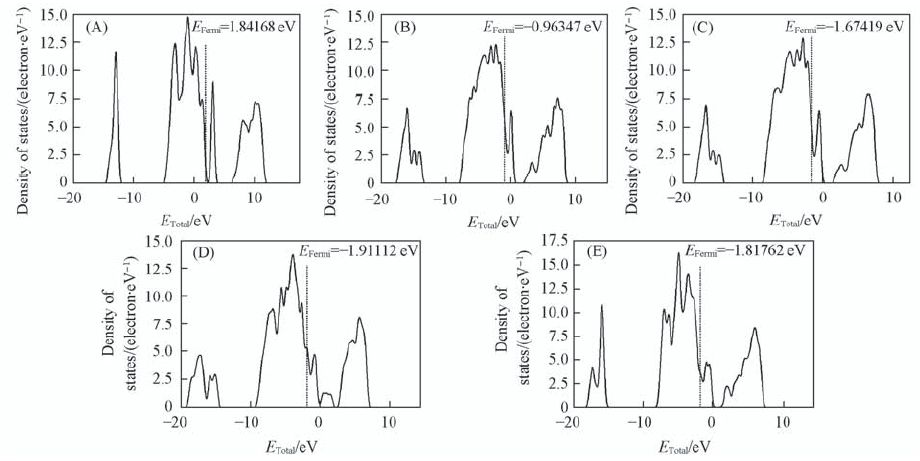
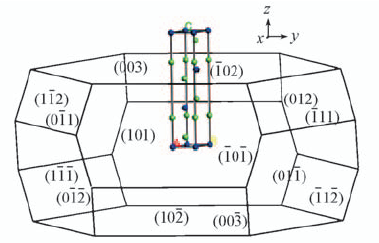
| [1] |
Prakash J., Redey L., Vissers D. R., Journal of Power Sources,1999, 84, 63—69
|
| [2] |
GuoY. Q., Wen J. F., Zhao J. F., Li X., Zhang W. H., Lu X. L., Zhu J. C., Chinese Journal of Power Sources, 2010, 33(2), 174—176
|
|
(郭永全, 闻俊锋, 赵晋峰, 李潇, 张卫红, 鹿学玲, 朱金城. 电源技术, 2010, 33(2), 174—176)
|
| [3] |
GuoY. Q., Zhao J. F., Li X., Lu X. L., Gao W. M., Zhu J. C., Chinese Journal of Power Sources, 2010, 34(6), 556—558
|
|
(郭永全, 赵晋峰, 李潇, 鹿学玲, 高文明, 朱金城. , 2010, 34电源技术, 2010, 34(6), 556—558)
|
| [4] |
Shi Z., Han S. J., Xu Z. Z., Xue J., Zhang M., Wang S. L., Preparation Method of High Purity Anhydrous Nickel Chloride, CP 201010564426, 2011-05-18
|
|
(时卓, 韩绍娟, 徐壮志, 薛健, 张明, 王世林. 高纯无水氯化镍的制备方法, CP 201010564426, 2011-05-18)
|
| [5] |
Xing J. C., Zhu Y. L., Jiao Q. J., Journal of New Materials for Electrochemical Systems, 2014, 17, 209—211
|
|
(邢家超, 朱艳丽, 焦清介. 电化学系统用新型材料杂志, 2014, 17, 209—211)
|
| [6] |
Gibbs J.W., On the Equilibrium of Heterogeneous Substances, Collected Works, Longmans and Green Co., New York, 1928
|
| [7] |
Wulff G., Krist Z., Mineral, 1901, 34, 449—530
|
| [8] |
Rtyson W., Miller W. A., Surf. Sci., 1997, 62, 67—276
|
| [9] |
Wan J., Fan Y. L., Gongeta1 D. W., Model. Simul. Mater. Sci. Eng., 1999, 7, 189—206
|
| [10] |
Ahmad E. A., Mallia G., Kramer D., Kucernaka A. R., Harrisonabd N. M., Journal of Materials Chemistry A, 2013, 1, 11152—11162
|
| [11] |
Zhu W. H., Jin H. M., Wu P., Liu H. L., Phys. Rev. B,2004, 70, 165419-1—5
|
| [12] |
Nakayama M., Hotta S., Nakamura T., Kasuga T., Journal of the Ceramic Society of Japan, 2013, 121(8), 611—613
|
| [13] |
Bruno M., Aquilano D., Pastero L., Prencipe M., Crystal Growth & Design,2008, 8(7), 2163—2170
|
| [14] |
Methfessel M., Henning D., ScheZer M., Phys. Rev. B,1992, 46, 4816—4829
|
| [15] |
Kollar J., Vitos L., Skriver H. L., Phys. Rev. B,1994, 49, 11288—11292
|
| [16] |
ZhangY. F., Li J. J., Ding K. N., Chen W. K., Zhou L. X., Chem. J. Chinese Universities,2003, 24(5), 863—867
|
|
(章永凡, 李俊篯, 丁开宁, 陈文凯, 周立新. 高等学校化学学报,2003, 24(5), 863—867)
|
| [17] |
Jia J. Q., Xie X. J., Liang Z. H., Zhang X. C., Fan C. M., Han P. D., Chem. J. Chinese Universities,2012, 33(5), 1050—1056
|
|
(贾金乾, 解学佳, 梁镇海, 张小超, 樊彩梅, 韩培德. 高等学校化学学报,2012, 33(5), 1050—1056)
|
| [18] |
Qi K. Z., Wang Y., Fu J. Q., Selvaraj R., Wang G. C., Chem. J. Chinese Universities,2014, 35(12), 2523—2528
|
|
(戚克振, 王艳, 付嘉琦, Selvaraj Rengaraj, 王贵昌. 高等学校化学学报,2014, 35(12), 2523—2528)
|
| [19] |
Donnay J. D. H., Harker D., Amer. Mineral,1937, 22, 446—447
|
| [20] |
Hartman P., Perdok W. G., Acta Crystallogr,1955, 8, 525—529
|
| [21] |
Wu Z. P., Yin Z. L., Chen Q. Y., Li J., Journal of Central South University(Science and Technology), 2009, 40(4), 897—903
|
|
(吴争平, 尹周澜, 陈启元, 李洁. 中南大学学报(自然科学版), 2009, 40(4), 897—903)
|
| [22] |
Segall M. D., Lindan P. J. D., Probert M. J., Pickard C., Hasnip P. J., Clark S. J., Payne M. C., J. Phys. Cond. Matt., 2002, 14(11), 2717—2743
|
| [23] |
Vanderbilt D., Phys. Rev. B, 1990, 41, 7892—7895
|
| [24] |
Casassa S., Pisani C., Phys. Rev. B,1995, 51, 7805—7816
|
| [25] |
Polatoglou H. M., Methfessel M., Phys. Rev. B,1993, 48, 1877—1883
|
| [26] |
Bravais A., Etudes Cristallographiques, Gauthier, Villars, 1866
|
| [27] |
Friedel G., Bull. Soc. Fr. Mineral, 1908, 30, 326—455
|
| [28] |
Ferrari A., Braibanti A., Bigliardi G., Acta Crystallogr,1963, 16, 846—847
|
 ), 赵勇, 邢家超, 焦清介
), 赵勇, 邢家超, 焦清介