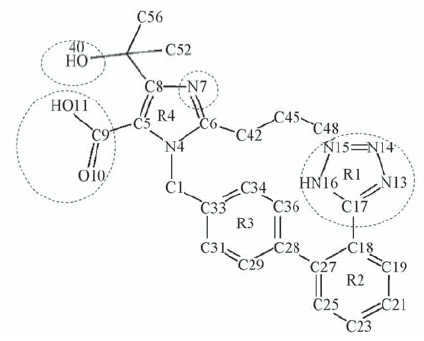
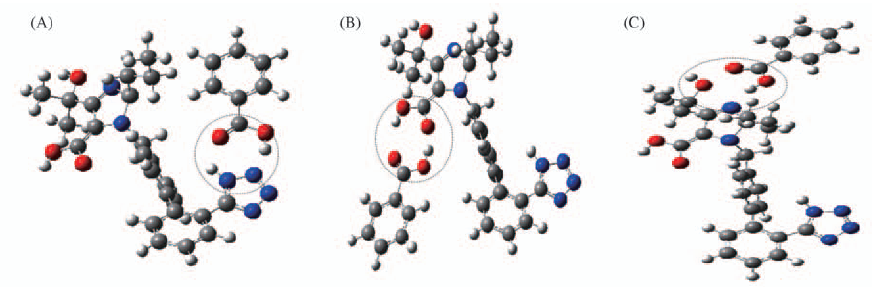
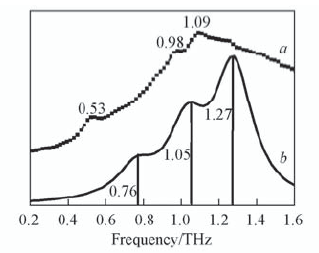
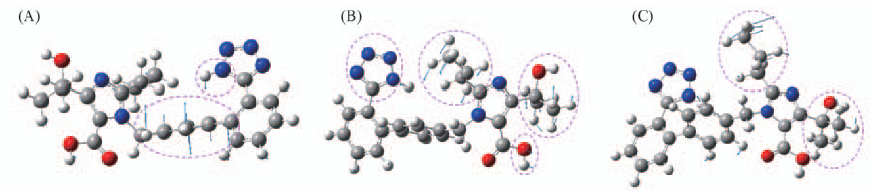
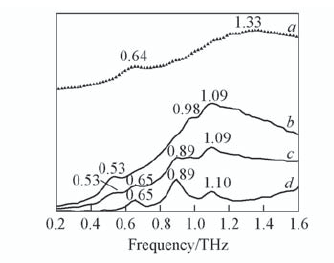
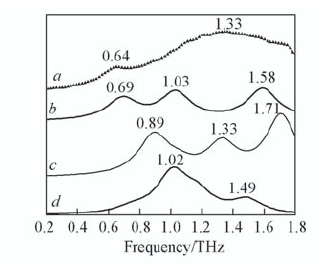
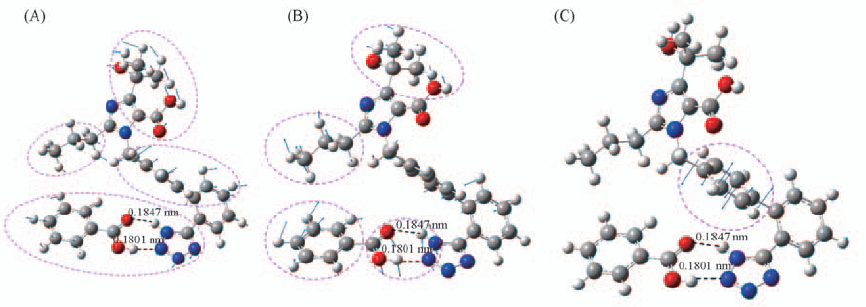
| [1] |
Lara O. F., Espinosa P. G., Supramol. Chem., 2007, 19(8), 553—557
|
| [2] |
Shah N., Zaworotko M., J. Drug Discov. Today,2008, 13(9/10), 440—446
|
| [3] |
Ji B. M., Deng D. S., Ma N., Miao S. B., Liu P., Li X. F., Chem. J. Chinese Universities,2011, 32(9), 2114—2122
|
|
(吉保明, 邓冬生, 马宁, 苗少斌, 刘鹏, 李贤飞. 高等学校化学学报,2011, 32(9), 2114—2122)
|
| [4] |
Walsh R.D. B., Bradner M.W., Fleischman S., Moralcs L. H., Mouhon B., Rodriguez H. N., Zaworotko M. J.,Chem. Commun., 2003, 186—187
|
| [5] |
Trask A., Motherwell W., Jones W., Int. J. Pharm., 2006, 320(1/2), 114—123
|
| [6] |
Nehm S., Rodriguez-Spong B., Rodriguez-Hornedo N., Cryst. Growth Des., 2006, 6(2), 592—600
|
| [7] |
Tanigawara Y., Yoshihara K., Kuramoto K., Arakawa K., Drug Metab. Pharmacokunet., 2009, 24(4), 376—378
|
| [8] |
Tanaka H., Nagasawa Y., Matsui I., Hamano T., Iwatani H., Kawada N., Horio M., Ito T., Isaka Y., Imai E., Clin. Exp. Nephrol., 2009, 13(1), 61—65
|
| [9] |
Tan Z. R., Li H., Ouyang D. S., Zhou H. H., Chinese New Drugs J., 2012, 21(6), 628—632
|
|
(谭志荣, 李慧, 欧阳冬生, 周宏灏. 中国新药杂志, 2012, 21(6), 628—632)
|
| [10] |
Laeis P., Puchler K., Kirch W., J. Hypertens., 2001, 19(S1), S21—S32
|
| [11] |
Chen J. M., Wu C. B., Lu T. B., Chem. J. Chinese Universities,2011, 32(9), 1999—2009
|
|
(陈嘉媚, 吴传斌, 鲁统部. 高等学校化学学报,2011, 32(9), 1999—2009)
|
| [12] |
Yi D. Y., Hong M H., Xu J., Zhao W. J., Ren G. B., Chinese J. Antibiotics,2011, 36(8), 561—575
|
|
(弋东阳, 洪鸣凰, 徐军, 赵文杰, 任国宾. 中国抗生素杂志,2011, 36(8), 561—575)
|
| [13] |
Gao Y., Zhu H., Zhang J., J. Prog. Chem., 2010, 22(5), 829—836
|
|
(高缘, 祖卉, 张建军. 化学进展, 2010, 22(5), 829—836)
|
| [14] |
Wang J. L., Jia X. H., Int. J. Pharm., 2007, 34(6), 434—438
|
|
(王京丽, 贾秀虹. 国际药学研究杂志, 2007, 34(6), 434—438)
|
| [15] |
Dubey R., Desiraju G. R., Chem. Commun., 2014, 50(10), 1181—1184
|
| [16] |
Osmialowski B., Kolehmainnen E., Ejsmont K., Ikonen S., Valkonen A., Rissanen K., Nonappa J. Mol. Struct., 2013, 1054, 157—163
|
| [17] |
Mazal W., Jole B., Angew. Chem. Int. Ed., 2006, 45, 7966—7969
|
| [18] |
Harry J. B., Cryst. Growth Des., 2009, 9(5), 2492—2499
|
| [19] |
Harry J. B., Cryst. Growth Des., 2009, 9(8), 3497—3503
|
| [20] |
Liu J. F., Wang J., Yu Y., Hou Y. N., Chinese J. Pharm. Anal., 2006, 26(5), 595—597
|
|
(刘建芳, 王金, 于洋, 侯艳宁. 药物分析杂志, 2006, 26(5), 595—597)
|
| [21] |
Yang W. Y., Li Z., Tang S. X., Ji Y. P., Kang X., Hu J. H., Pharm. Care Res., 2009, 9(5), 367—369
|
|
(杨武云, 李珍, 唐世新, 计一平, 康新, 胡晋红. 药物服务与研究, 2009, 9(5), 367—369)
|
| [22] |
Zhang X. L., Yu Y., Hou Y. N., Zhao H. Q., Chinese J. Hosp. Pharm., 2007, 27(5), 622—624
|
|
(张小丽, 于洋, 侯艳红, 赵怀清. 中国医院药学杂志, 2007, 27(5), 622—624)
|
| [23] |
Vikas V., Shikha M., Chromatographia., 2008, 67(1/2), 147—150
|
| [24] |
Liu D. Y., Hu P., Matsushima N., Li X. M., Li L., Jiang L., J. Chromato. B,2007, 856(1/2), 190—197
|
| [25] |
Carlor A. F., Susana B. E., Reinaldo P. D., Patricia A. M. W., J. Raman Spectrosc., 2009, 40, 1296—1300
|
| [26] |
Rong Y. Z., Zhao B., Huang X. H., Wu Y., J. Nanjing Normal University(Natural Science Edition), 2008, 31(1), 71—74
|
|
(荣玉芝, 赵波, 黄晓华, 吴芸. 南京师大学报(自然科学版), 2008, 31(1), 71—74)
|
| [27] |
Sean P. D., Duohai P., Michael G., Shawn X. Y., Timothy M. K., Cryst. Growth Des., 2012, 12, 5017—5024
|
| [28] |
Xu J.Z., Zhang X. C., Technology and Applications of Terahertz Wave, Peking University Press, Beijing, 2007, 1—5
|
|
(许景周, 张希成. 太赫兹波科学技术与应用, 北京: 北京大学出版社, 2007, 1—5)
|
| [29] |
Fang H. X., Zhang Q., Zhang H. L., Du Y., Hong Z., Acta Phys.-Chim. Sinica,2015, 31(2), 221—226
|
|
(方虹霞, 张琪, 张慧丽, 杜勇, 洪治. 物理化学学报,2015, 31(2), 221—226)
|
| [30] |
Du Y., Xia Y., Zhang H. L., Hong Z., Spectrochim Acta A,2013, 111,192—195
|
| [31] |
Nguyen K. L., Friscic T., Day G. M., Gladden L. F., Jones W., Nat. Mater., 2009, 6(3), 206—209
|
| [32] |
Yang C. Q., Wang J., Zhang Z. W., Spectrosc. Spec. Anal., 2011, 31(9), 2476—2479
|
|
(杨彩霞, 王静, 张振伟. 光谱学与光谱分析, 2011, 31(9), 2476—2479)
|
| [33] |
Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A. Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A.G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Gaussian 03, Revision E.01, Gaussian Inc., Wallingford CT, 2004
|
| [34] |
King M. D., Buchanan W. D., Korter T. M., Anal. Chem., 2011, 83(10), 3786—3792
|
| [35] |
Hakey P. M., Allis D. G., Ouellette W., Korter T. M., J. Phys. Chem. A,2009, 113(10), 5119—5127
|
| [36] |
King M. D., Buchanan W. D., Korter T. M., Phys. Chem. Chem. Phys., 2011, 13(10), 4250—4259
|
| [37] |
Perumalla S. R., Suresh E., Pedireddi V. R., Angew. Chem. Int. Ed., 2005, 44, 7752—7757
|
| [38] |
Zheng Z. P., Fan W. H., Yan H., Liu J., Xu L. M., Spectrosc. Spec. Anal., 2013, 33(3), 582—585
|
|
(郑转平, 范文慧, 闫慧, 刘佳, 徐利民. 光谱学与光谱分析, 2013, 33(3), 582—585)
|
 )
)