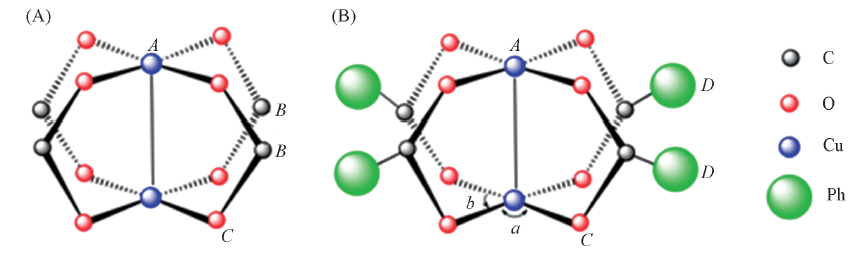
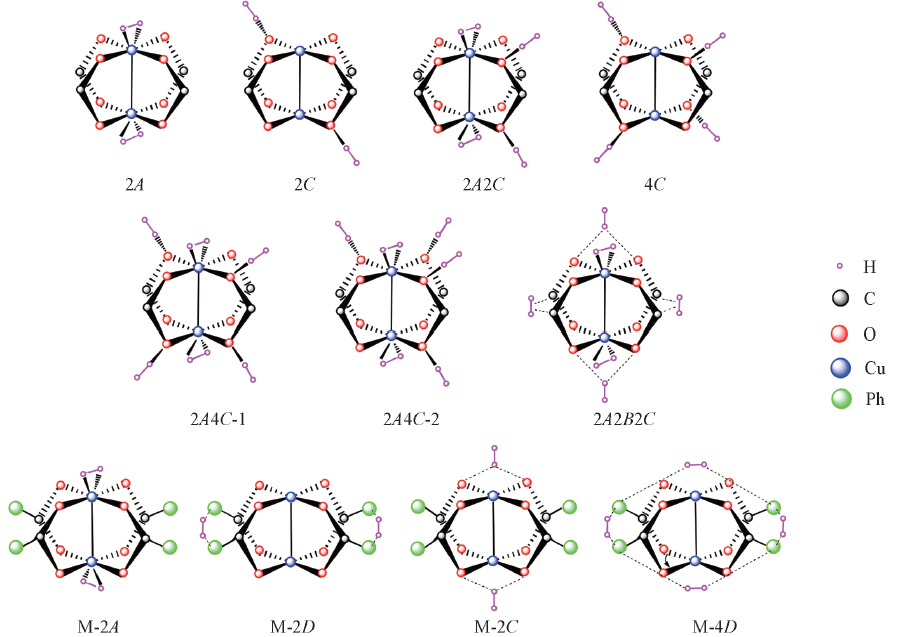
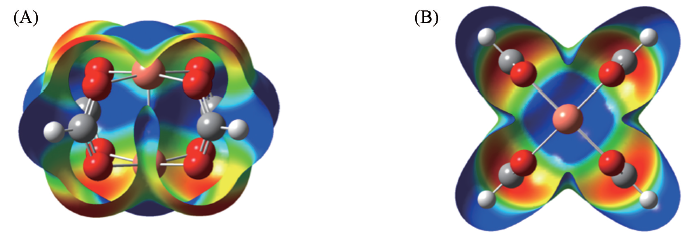
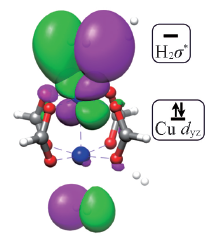
| [1] |
Li J. L., Zhang X., Huang X. R., Phys. Chem. Chem. Phys., 2012, 14, 246—256
|
| [2] |
Sun X. L., Huang X. R., Li J. L., Huo R. P., Sun C. C., J. Phys. Chem. A, 2012, 116, 1475—1485
|
| [3] |
Huo R. P., Zhang X., Huang X. R., Li J. L., Sun C. C., Acta Chim. Sinica, 2013, 71, 743—748
|
|
(霍瑞萍, 张祥, 黄旭日, 李吉来, 孙家锺. 化学学报, 2013, 71, 743—748)
|
| [4] |
Li J. L., Mata R. A., Ryde U., J. Chem. Theory. Comput., 2013, 9, 1799—1807
|
| [5] |
Shi Q., Bergquist K. E., Huo R. P., Li J. L., Lund M., Vacha R., Sundin A., Butkus E., Orentas E., Warnmark K., J. Am. Chem. Soc., 2013, 135, 15263—15268
|
| [6] |
Sun X. L., Li J. L., Huang X. R., Sun C. C., Acta Chim. Sinica, 2013, 71, 749—754
|
|
(孙小丽, 李吉来, 黄旭日, 孙家锺. 化学学报, 2013, 71, 749—754)
|
| [7] |
Li J. L., Geng C. Y., Bu Y. X., Huang X. R., Sun C. C., J. Comput. Chem., 2009, 30, 1135—1145
|
| [8] |
Li J. L., Geng C. Y., Huang X. R., Sun C. C., J. Chem. Theory. Comput., 2006, 2, 1551—1564
|
| [9] |
Li J. L., Geng C. Y., Huang X. R., Zhang X., Sun C. C., Organometallics, 2007, 26, 2203—2210
|
| [10] |
Geng C. Y., Li J. L., Huang X. R., Liu H. L., Li Z., Sun C. C., J. Comput. Chem., 2008, 29, 686—693
|
| [11] |
Geng C.Y., Li J. L., Huang X. R., Sun C. C., Chem. Phys., 2006, 324, 474—482
|
| [12] |
Roy D., Marianski M., Maitra N. T., Dannenberg J. J., J. Chem. Phys., 2012, 137, 134109
|
| [13] |
Ess D. H., Houk K. N., J. Phys. Chem. A, 2005, 109, 9542—9553
|
| [14] |
Sun X. H., Sun X. L., Geng C. Y., Zhao H. T., Li J. L., J. Phys. Chem. A, 2014, 118, 7146—7158
|
| [15] |
Andreji M., Mata R. A., J. Chem.Theory. Comput., 2014, 10, 5397—5404
|
| [16] |
Zhang X., Schwarz H., Theor. Chem. Acc., 2011, 129, 389—399
|
| [17] |
Korth M., Grimme S., J. Chem. Theory. Comput., 2009, 5, 993—1003
|
| [18] |
Li J. L., González-Navarrete P., Schlangen M., Schwarz H., Chem. Eur. J., 2015, 21, 7780—7789
|
| [19] |
Cohen A. J., Mori-Sánchez P., Yang W., Chem. Rev., 2012, 112, 289—320
|
| [20] |
Boguslawski K., Jacob C. R., Reiher M., J. Chem. Theory. Comput., 2011, 7, 2740—2752
|
| [21] |
Yu Y. X., ACS Appl. Mater. Interfaces, 2014, 6, 16267—16275
|
| [22] |
Yu Y. X., J. Mater. Chem. A, 2014, 2, 8910—8917
|
| [23] |
Wang C.J., Tang C. M., Zhang Y. J., Gao F. Z., Chem. J. Chinese Universities, 2014, 35,2131—2137
|
|
(王成杰, 唐春梅, 张轶杰, 高凤志. 高等学校化学学报, 2014, 35,2131—2137)
|
| [24] |
Grimme S., Chem. Eur. J., 2012, 18, 9955—9964
|
| [25] |
Grimme S., J. Comput. Chem., 2006, 27, 1787—1799
|
| [26] |
Goerigk L., Grimme S., Phys. Chem. Chem. Phys., 2011, 13, 6670—6688
|
| [27] |
Korth M., Grimme S., J. Chem. Theory Comput., 2009, 5, 993—1003
|
| [28] |
Wang S. Y., Zhong C. L., Acta Chim. Sinica, 2006, 64, 2375—2378
|
|
(王三跃, 仲崇立. 化学学报, 2006, 64, 2375—2378)
|
| [29] |
Li J. L., Geng C. Y., Huang X. R., Sun C. C., J. Chem. Theory Comput., 2006, 2, 1551—1564
|
| [30] |
Toda J., Fischer M., Jorge M., Gomes J. R. B., Chem. Phys. Lett., 2013, 587, 7—13
|
| [31] |
Grajciar L., Nachtigall P., Bludsk O., Rubeš M., J. Chem. Theory Comput., 2015, 11, 230—238
|
| [32] |
Terencio T., Di Renzo F., Berthomieu D., Trens P., J. Phys. Chem. C, 2013, 117, 26156—26165
|
| [33] |
Brittain D. R. B., Lin C. Y., Gilbert A. T. B., Izgorodina E. I., Gill P. M. W., Coote M. L., Phys. Chem. Chem. Phys., 2009, 11, 1138—1142
|
| [34] |
Sun X. L., Geng C. Y., Huo R. P., Ryde U., Bu Y. X., Li J. L., J. Phys. Chem. B, 2014, 118, 1493—1500
|
| [35] |
Huo R. P., Zhang X., Huang X. R., Li J. L., Sun C. C., J. Phys. Chem. A, 2011, 115, 3576—3582
|
| [36] |
Li N., Huo R. P., Zhang X., Huang X. R., Li J. L., Sun C. C., Chem. Phys. Lett., 2011, 503, 210—214
|
| [37] |
Dewar M., Bull. Soc. Chem. Fr., 1951, 18, C79
|
| [38] |
Chatt J., Duncanson L.A.,J. Chem. Soc., 1953, 2939
|
| [39] |
Chatt J., Duncanson L.A., Venanzi M. L.,J. Chem. Soc., 1955, 4456—4460
|
| [40] |
Bernardo C. P., Bauman N., Piecuch P., Silva P., J. Mol. Model., 2013, 19, 5457—5467
|
| [41] |
Zhao Y., Truhlar D. G., J. Phys. Chem. A, 2005, 109, 5656—5667
|
| [42] |
Hirscher M., Panella B., Schmitz B., Microporous. Mesoporous. Mater., 2010, 129, 335—339
|
 )
)