

高等学校化学学报 ›› 2014, Vol. 35 ›› Issue (5): 1000.doi: 10.7503/cjcu20140060
夏喜泉, 张辉, 张桂玲
收稿日期:2014-01-20
出版日期:2014-05-10
发布日期:2014-04-29
作者简介:联系人简介: 张桂玲, 女, 教授, 主要从事光电磁材料的理论及实验研究. E-mail:基金资助:
XIA Xiquan, ZHANG Hui, ZHANG Guiling*( )
)
Received:2014-01-20
Online:2014-05-10
Published:2014-04-29
Contact:
ZHANG Guiling
E-mail:1621717290@qq.com
Supported by:摘要:
以N749染料为母体, 保留三联吡啶配体(tcterpy)作为辅助配体, 利用两齿的N-杂环卡宾-吡啶配体(NHC-py)替代2个硫氰酸(NCS)配体设计了一系列同时含有三齿配体和两齿配体的染料分子1~4. 利用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)方法对染料分子1~4及母体分子N749的几何结构、 电子结构和光谱性质进行了系统的理论研究. 研究结果表明, 该系列分子具有良好的光吸收性能, 最低能吸收波长可达到800 nm, 吸收跃迁为MLCT/LLCT混合跃迁.
中图分类号:
TrendMD:
夏喜泉, 张辉, 张桂玲. N-杂环卡宾-吡啶基钌光敏染料的电子结构和光谱性质的理论研究. 高等学校化学学报, 2014, 35(5): 1000.
XIA Xiquan, ZHANG Hui, ZHANG Guiling. Theoretical Studies on the Structures and Spectroscopic Properties of N-Heterocyclic Carbene-pyridine-based Ruthenium Sensitizers†. Chem. J. Chinese Universities, 2014, 35(5): 1000.
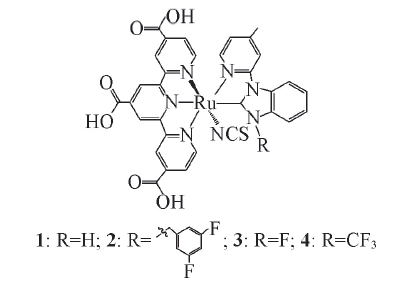
Fig.1 Schematic representation of complexes 1, 2, 3 and 4
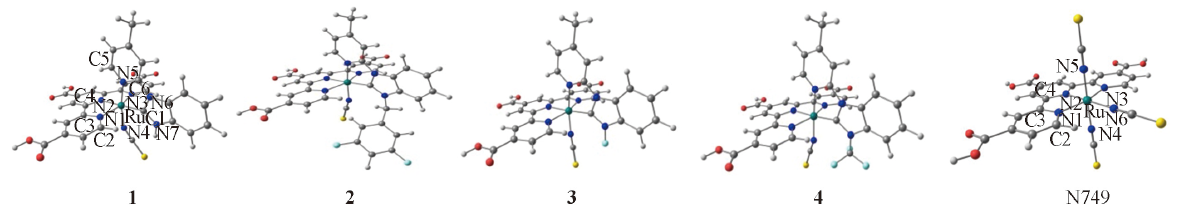
Fig.2 Optimized geometry structures of complexes 1—4 and N749 at B3LYP/LanL2DZ level of theory
| Parameter | 1 | 2 | 3 | 4 | N749 | Expt.[ |
|---|---|---|---|---|---|---|
| Ru—N1/nm | 0.2098 | 0.2106 | 0.2099 | 0.2101 | 0.2062 | 0.2090 |
| Ru—N2/nm | 0.2022 | 0.2024 | 0.2018 | 0.2026 | 0.1937 | 0.1936 |
| Ru—N3/nm | 0.2097 | 0.2095 | 0.2098 | 0.2100 | 0.2062 | |
| Ru—N4/nm | 0.2066 | 0.2040 | 0.2030 | 0.2040 | 0.2040 | 0.2032 |
| Ru—N5/nm | 0.2108 | 0.2102 | 0.2117 | 0.2101 | 0.2040 | |
| Ru—C1(N6)/nm | 0.2006 | 0.2029 | 0.2006 | 0.2019 | 0.2073 | 0.2052 |
| N1—C2/nm | 0.1361 | 0.1360 | 0.1360 | 0.1359 | 0.1358 | |
| N1—C3/nm | 0.1387 | 0.1387 | 0.1387 | 0.1386 | 0.1389 | |
| N2—C4/nm | 0.1367 | 0.1365 | 0.1367 | 0.1364 | 0.1379 | |
| N5—C5/nm | 0.1363 | 0.1363 | 0.1364 | 0.1364 | ||
| N5—C6/nm | 0.1375 | 0.1371 | 0.1372 | 0.1369 | ||
| N1—Ru—N2/(°) | 78.8 | 78.7 | 78.9 | 78.8 | 80.5 | 81.1 |
| N3—Ru—N2/(°) | 78.8 | 78.8 | 78.8 | 78.7 | 80.5 | |
| N1—Ru—N3/(°) | 157.5 | 157.6 | 157.6 | 157.4 | 161.1 | 161.6 |
| N5—Ru—C1/(°) | 77.6 | 77.9 | 77.2 | 77.6 |
Table 1 Partial optimized geometry structural parameters of complexes 1—4 and N749 in the ground states using the DFT methods and the experimental data of N749
| Parameter | 1 | 2 | 3 | 4 | N749 | Expt.[ |
|---|---|---|---|---|---|---|
| Ru—N1/nm | 0.2098 | 0.2106 | 0.2099 | 0.2101 | 0.2062 | 0.2090 |
| Ru—N2/nm | 0.2022 | 0.2024 | 0.2018 | 0.2026 | 0.1937 | 0.1936 |
| Ru—N3/nm | 0.2097 | 0.2095 | 0.2098 | 0.2100 | 0.2062 | |
| Ru—N4/nm | 0.2066 | 0.2040 | 0.2030 | 0.2040 | 0.2040 | 0.2032 |
| Ru—N5/nm | 0.2108 | 0.2102 | 0.2117 | 0.2101 | 0.2040 | |
| Ru—C1(N6)/nm | 0.2006 | 0.2029 | 0.2006 | 0.2019 | 0.2073 | 0.2052 |
| N1—C2/nm | 0.1361 | 0.1360 | 0.1360 | 0.1359 | 0.1358 | |
| N1—C3/nm | 0.1387 | 0.1387 | 0.1387 | 0.1386 | 0.1389 | |
| N2—C4/nm | 0.1367 | 0.1365 | 0.1367 | 0.1364 | 0.1379 | |
| N5—C5/nm | 0.1363 | 0.1363 | 0.1364 | 0.1364 | ||
| N5—C6/nm | 0.1375 | 0.1371 | 0.1372 | 0.1369 | ||
| N1—Ru—N2/(°) | 78.8 | 78.7 | 78.9 | 78.8 | 80.5 | 81.1 |
| N3—Ru—N2/(°) | 78.8 | 78.8 | 78.8 | 78.7 | 80.5 | |
| N1—Ru—N3/(°) | 157.5 | 157.6 | 157.6 | 157.4 | 161.1 | 161.6 |
| N5—Ru—C1/(°) | 77.6 | 77.9 | 77.2 | 77.6 |
| MO | Energy/eV | Compositions(%) | Assignment of orbital | |||
|---|---|---|---|---|---|---|
| Ru | tcterpy | NHC-py | NCS | |||
| LUMO+7 | -1.35159 | 1 | 0 | 98 | 0 | π*(NHC-py) |
| LUMO+4 | -2.12167 | 8 | 2 | 91 | 1 | π*(NHC-py) |
| LUMO+3 | -2.57447 | 1 | 99 | 1 | 0 | π*(tcterpy) |
| LUMO+2 | -2.80957 | 1 | 99 | 0 | 0 | π*(tcterpy) |
| LUMO+1 | -3.08523 | 5 | 94 | 0 | 0 | π*(tcterpy) |
| LUMO | -3.52687 | 7 | 92 | 0 | 1 | π*(tcterpy) |
| HOMO | -5.83602 | 32 | 5 | 4 | 58 | d(Ru)-π*(NCS) |
| HOMO-1 | -5.89943 | 24 | 6 | 1 | 68 | d(Ru)-π*(NCS) |
| HOMO-2 | -6.55658 | 55 | 7 | 38 | 1 | d(Ru)-π*(NHC-py) |
| HOMO-3 | -6.87142 | 36 | 13 | 9 | 41 | d(Ru)-π*(NCS) |
| HOMO-4 | -7.09863 | 48 | 15 | 2 | 35 | d(Ru)-π*(NCS) |
| HOMO-5 | -7.24748 | 3 | 3 | 95 | 1 | π(NHC-py) |
| HOMO-6 | -7.69565 | 2 | 95 | 1 | 1 | π(tcterpy) |
| HOMO-7 | -7.86817 | 18 | 4 | 76 | 1 | π(NHC-py) |
| HOMO-8 | -8.17756 | 0 | 0 | 100 | 0 | π(NHC-py) |
| HOMO-9 | -8.49675 | 2 | 96 | 0 | 1 | π(tcterpy) |
| HOMO-10 | -8.59227 | 1 | 89 | 10 | 0 | p(COOH) |
Table 2 Partial molecular orbital compositions of complex 3 in CH3CN under the TD-DFT calculations
| MO | Energy/eV | Compositions(%) | Assignment of orbital | |||
|---|---|---|---|---|---|---|
| Ru | tcterpy | NHC-py | NCS | |||
| LUMO+7 | -1.35159 | 1 | 0 | 98 | 0 | π*(NHC-py) |
| LUMO+4 | -2.12167 | 8 | 2 | 91 | 1 | π*(NHC-py) |
| LUMO+3 | -2.57447 | 1 | 99 | 1 | 0 | π*(tcterpy) |
| LUMO+2 | -2.80957 | 1 | 99 | 0 | 0 | π*(tcterpy) |
| LUMO+1 | -3.08523 | 5 | 94 | 0 | 0 | π*(tcterpy) |
| LUMO | -3.52687 | 7 | 92 | 0 | 1 | π*(tcterpy) |
| HOMO | -5.83602 | 32 | 5 | 4 | 58 | d(Ru)-π*(NCS) |
| HOMO-1 | -5.89943 | 24 | 6 | 1 | 68 | d(Ru)-π*(NCS) |
| HOMO-2 | -6.55658 | 55 | 7 | 38 | 1 | d(Ru)-π*(NHC-py) |
| HOMO-3 | -6.87142 | 36 | 13 | 9 | 41 | d(Ru)-π*(NCS) |
| HOMO-4 | -7.09863 | 48 | 15 | 2 | 35 | d(Ru)-π*(NCS) |
| HOMO-5 | -7.24748 | 3 | 3 | 95 | 1 | π(NHC-py) |
| HOMO-6 | -7.69565 | 2 | 95 | 1 | 1 | π(tcterpy) |
| HOMO-7 | -7.86817 | 18 | 4 | 76 | 1 | π(NHC-py) |
| HOMO-8 | -8.17756 | 0 | 0 | 100 | 0 | π(NHC-py) |
| HOMO-9 | -8.49675 | 2 | 96 | 0 | 1 | π(tcterpy) |
| HOMO-10 | -8.59227 | 1 | 89 | 10 | 0 | p(COOH) |
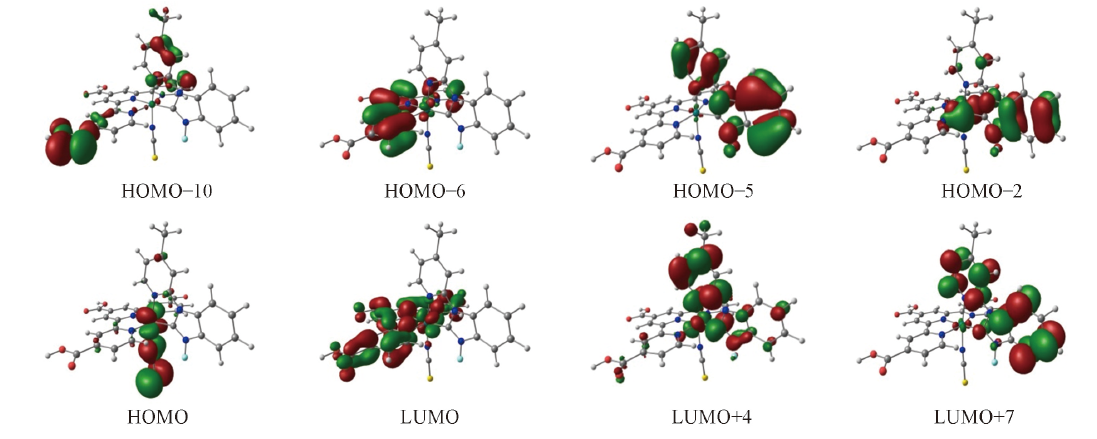
Fig.3 Electron density diagrams of the frontier molecular orbitals relevant to the absportions of complex 3 under the TD-DFT calculations
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A 1A | H→L(0.69) | 729(1.70) | 0.0329 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.61) | 610(2.03) | 0.0310 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 585(2.12) | 0.0028 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.65) | 521(2.38) | 0.0665 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.68) | 477(2.60) | 0.1015 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.63) | 370(3.35) | 0.1452 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.54) | 336(3.69) | 0.4367 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.39) | MLCT/LLCT | |||||||||
| H 1A | H-4→L+4(0.45) | 266(4.66) | 0.3790 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.28) | MLCT/LLCT | |||||||||
| 2 | A 1A | H→L(0.69) | 769(1.61) | 0.0309 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.66) | 660(1.88) | 0.0156 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 594(2.09) | 0.0017 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.69) | 546(2.27) | 0.0326 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 495(2.50) | 0.0803 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.59) | 380(3.26) | 0.1104 | MLCT/LLCT | ||||||
| G 1A | H-8→L(0.52) | 339(3.66) | 0.2986 | π(tcterpy)→π*(tcterpy) | ||||||
| H 1A | H-5→L+4(0.52) | 280(4.43) | 0.0955 | π(NHC-py)→π*(NHC-py) | ||||||
| 3 | A 1A | H→L(0.69) | 773(1.60) | 0.0259 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.68) | 683(1.82) | 0.0259 | MLCT/LLCT | ||||||
| C 1A | H-1→L+1(0.70) | 565(2.20) | 0.0266 | MLCT/LLCT | ||||||
| D 1A | H-2→L(0.69) | 557(2.23) | 0.0018 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 480(2.58) | 0.0342 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.61) | 378(3.28) | 0.1665 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.59) | 340(3.65) | 0.4973 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.31) | MLCT/LLCT | |||||||||
| H 1A | H-5→L+4(0.52) | 277(4.48) | 0.3485 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.37) | MLCT | |||||||||
| 4 | A 1A | H→L(0.70) | 777(1.60) | 0.0243 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.69) | 693(1.79) | 0.0136 | MLCT/LLCT | ||||||
| C 1A | H→L+1(0.67) | 564(2.20) | 0.0165 | MLCT/LLCT | ||||||
| D 1A | H→L+2(0.69) | 498(2.49) | 0.0511 | MLCT/LLCT | ||||||
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
| 4 | E 1A | H-3→L+1(0.61) | 382(3.24) | 0.1711 | MLCT/LLCT | |||||
| F 1A | H-6→L(0.59) | 340(3.64) | 0.5049 | π(tcterpy)→π*(tcterpy) | ||||||
| G 1A | H-5→L+4(0.58) | 275(4.50) | 0.0790 | π(NHC-py)→π*(NHC-py) | ||||||
| N749 | A 1A' | H→L+1(0.65) | 859(1.44) | 0.0409 | MLCT/LLCT | |||||
| B 1A' | H→L+2(0.57) | 686(1.80) | 0.1182 | MLCT/LLCT | 625 | |||||
| C 1A″ | H-6→L(0.66) | 513(2.42) | 0.0516 | MLCT/LLCT | 556 | |||||
| D 1A' | H-6→L+2(0.64) | 393(3.16) | 0.1586 | MLCT/LLCT | 429 | |||||
| E 1A' | H-9→L(0.64) | 334(3.72) | 0.2683 | π(tcterpy)→π*(tcterpy) | 344,330 | |||||
Table 3 Calculated absorptions of complexes 1—4 and N749 in CH3CN at TD-DFT(B3LYP) level*
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A 1A | H→L(0.69) | 729(1.70) | 0.0329 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.61) | 610(2.03) | 0.0310 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 585(2.12) | 0.0028 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.65) | 521(2.38) | 0.0665 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.68) | 477(2.60) | 0.1015 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.63) | 370(3.35) | 0.1452 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.54) | 336(3.69) | 0.4367 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.39) | MLCT/LLCT | |||||||||
| H 1A | H-4→L+4(0.45) | 266(4.66) | 0.3790 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.28) | MLCT/LLCT | |||||||||
| 2 | A 1A | H→L(0.69) | 769(1.61) | 0.0309 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.66) | 660(1.88) | 0.0156 | MLCT/LLCT | ||||||
| C 1A | H-2→L(0.69) | 594(2.09) | 0.0017 | MLCT/LLCT | ||||||
| D 1A | H-1→L+1(0.69) | 546(2.27) | 0.0326 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 495(2.50) | 0.0803 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.59) | 380(3.26) | 0.1104 | MLCT/LLCT | ||||||
| G 1A | H-8→L(0.52) | 339(3.66) | 0.2986 | π(tcterpy)→π*(tcterpy) | ||||||
| H 1A | H-5→L+4(0.52) | 280(4.43) | 0.0955 | π(NHC-py)→π*(NHC-py) | ||||||
| 3 | A 1A | H→L(0.69) | 773(1.60) | 0.0259 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.68) | 683(1.82) | 0.0259 | MLCT/LLCT | ||||||
| C 1A | H-1→L+1(0.70) | 565(2.20) | 0.0266 | MLCT/LLCT | ||||||
| D 1A | H-2→L(0.69) | 557(2.23) | 0.0018 | MLCT/LLCT | ||||||
| E 1A | H→L+2(0.69) | 480(2.58) | 0.0342 | MLCT/LLCT | ||||||
| F 1A | H-3→L+1(0.61) | 378(3.28) | 0.1665 | MLCT/LLCT | ||||||
| G 1A | H-6→L(0.59) | 340(3.65) | 0.4973 | π(tcterpy)→π*(tcterpy) | ||||||
| H-3→L+2(0.31) | MLCT/LLCT | |||||||||
| H 1A | H-5→L+4(0.52) | 277(4.48) | 0.3485 | π(NHC-py)→π*(NHC-py) | ||||||
| H-2→L+7(0.37) | MLCT | |||||||||
| 4 | A 1A | H→L(0.70) | 777(1.60) | 0.0243 | MLCT/LLCT | |||||
| B 1A | H-1→L(0.69) | 693(1.79) | 0.0136 | MLCT/LLCT | ||||||
| C 1A | H→L+1(0.67) | 564(2.20) | 0.0165 | MLCT/LLCT | ||||||
| D 1A | H→L+2(0.69) | 498(2.49) | 0.0511 | MLCT/LLCT | ||||||
| Complex | State | Config(|CI| coef.) | λ/nm(E/eV) | Oscillator strength | Assignment | Expt. [ | ||||
| 4 | E 1A | H-3→L+1(0.61) | 382(3.24) | 0.1711 | MLCT/LLCT | |||||
| F 1A | H-6→L(0.59) | 340(3.64) | 0.5049 | π(tcterpy)→π*(tcterpy) | ||||||
| G 1A | H-5→L+4(0.58) | 275(4.50) | 0.0790 | π(NHC-py)→π*(NHC-py) | ||||||
| N749 | A 1A' | H→L+1(0.65) | 859(1.44) | 0.0409 | MLCT/LLCT | |||||
| B 1A' | H→L+2(0.57) | 686(1.80) | 0.1182 | MLCT/LLCT | 625 | |||||
| C 1A″ | H-6→L(0.66) | 513(2.42) | 0.0516 | MLCT/LLCT | 556 | |||||
| D 1A' | H-6→L+2(0.64) | 393(3.16) | 0.1586 | MLCT/LLCT | 429 | |||||
| E 1A' | H-9→L(0.64) | 334(3.72) | 0.2683 | π(tcterpy)→π*(tcterpy) | 344,330 | |||||
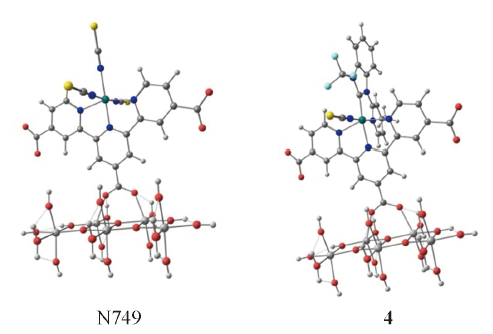
Fig.4 Optimized adsorption structure between N749, 4 and TiO2(101) surface
| [1] | O’Regan B., Grätzel M., Nature, 1991, 353, 737—740 |
| [2] | Snaith H. J., Adv. Funct. Mater., 2010, 20, 13—19 |
| [3] | Nazeeruddin M. K., Klein C., Liska P., Grätzel M., Coord. Chem. Rev., 2005, 249, 1460—1467 |
| [4] | Gao F., Wang Y., Shi D., Zhang J., Wang M., Jing X., Humphry-Baker R., Wang P., Zakeeruddin S. M., Grätzel M., J. Am. Chem. Soc., 2008, 130, 10720—10728 |
| [5] | Yum J. H., Jung I., Baik C., Ko J., Nazeeruddin M. K., Grätzel M., Energy Environ. Sci., 2009, 2, 100—102 |
| [6] | O’Regan B. C., Walley K., Juozapavicius M., Anderson A., Mater F., Ghaddar T., Zakeeruddin S. M., Klein C., Durrant J. R., J. Am. Chem. Soc., 2009, 131, 3541—3548 |
| [7] | Hagfeldt A., Boschloo G., Sun L.C., Klooand L., Pettersson H., Chem. Rev., 2010, 110, 6595—6663 |
| [8] | Zhang J., Li H. B., Wu Y., Geng Y., Duan Y. A., Liao Y., Su Z. M., Chem. J. Chinese Universities, 2011, 32(6), 1343—1348 |
| (张吉, 李海斌, 吴勇, 耿允, 段雨爱, 廖奕, 苏忠民.高等学校化学学报, 2011,32(6), 1343—1348) | |
| [9] | Nazeeruddin M. K., Kay A., Rodicio I., Humphry-Baker R., Muller E., Liska P., Vlachopoulos N., Grätzel M., J. Am. Chem. Soc., 1993, 115, 6382—6390 |
| [10] | Nazeeruddin M. K., DeAngelis F., Fantacci S., Selloni A., Viscardi G., Liska P., Ito S., Takeru B., Grätzel M., J. Am .Chem. Soc., 2005, 127, 16835—16847 |
| [11] | Nazeeruddin M.K., Pechy P., Grätzel M.,Chem. Commun., 1997, 1705—1706 |
| [12] | Nazeeruddin M. K., Pechy P., Renouard T., Zakeeruddin S. M., Humphry-Baker R., Comte P., Liska P., Cevey L., Costa E., Shklover V., Spiccia L., Deacon G. B., Bignozzi C. A., Grätzel M., J. Am. Chem. Soc., 2001, 123, 1613—1624 |
| [13] | Yu Q., Wang Y., Yi Z., Zu N., Zhang J., Zhang M., Wang P., ACS Nano, 2010, 4, 6032—6038 |
| [14] | Asghar M. I., Miettunen K., Halme J., Vahermaa P., Toivola M., Aitolaand K., Lund P., Energy Environ. Sci., 2010, 3, 418—426 |
| [15] | Wadman S.H., Kroon J. M., Bakker K., Lutz M., Spek A. L., van Klink G. P. M., van Koten G.,Chem. Commun., 2007, 1907—1909 |
| [16] | Bessho T., Yoneda E., Yum J. H., Guglielmi M., Tavernelli I., Imai H., Rothlisberger U., Nazeeruddin M. K., Grätzel M., J. Am. Chem. Soc., 2009, 131, 5930—5934 |
| [17] | Koivisto B. D., Robson K. C. D., Berlinguette C. P., Inorg. Chem., 2009, 48, 9644—9652 |
| [18] | Bomben P. G., Robson K. C. D., Koivisto B. D., Berlinguette C. P., Coord. Chem. Rev., 2012, 256, 1438—1450 |
| [19] | Brown D. G., Schauer P. A., Borau-Garcia J., Fancy B. R., Berlinguette C. P., J. Am. Chem. Soc., 2013, 135, 1692—1695 |
| [20] | Chou C.C., Wu K. L., Chi Y., Hu W. P., Yu S. J., Lee G. H., Lin C. L., Chou P. T., Angew. Chem. Int. Ed., 2011, 50, 2054—2058 |
| [21] | Chang W. C., Chen H. S., Li T. Y., Hsu N. M., Tingare Y. S., Li C. Y., Liu Y. C., Su. C. C., Li W. R., Angew. Chem. Int. Ed., 2010, 49, 8161—8164 |
| [22] | Su X., Zhang J., Wu Y., Geng Y., Su Z. M., Chem. J. Chinese Universities, 2013, 34(8), 1945—1952 |
| (苏欣, 张吉, 吴勇, 耿允, 苏忠民.高等学校化学学报, 2013,34(8), 1945—1952) | |
| [23] | Li C. M., Kan Y. H., Xu Y. Y., Duan Y. A., Su Z. M., Chem. J. Chinese Universities, 2012, 33(3), 591—597 |
| (李春敏, 阚玉和, 徐莹莹, 段雨爱, 苏忠民.高等学校化学学报, 2012,33(3), 591—597) | |
| [24] | Becke A. D., Chem. Phys., 1993, 98, 5648—5652 |
| [25] | Casida M. E., Jamorski C., Casida K. C., J. Chem. Phys., 1998, 108, 4439—4449 |
| [26] | Hay P. J., Wadt W. R., J. Chem. Phys., 1985, 82, 299—310 |
| [27] | Frisch M.J., Trucks G. W., Schlegel H. B. Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Toyota M. E. K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Peralta J. E. Jr., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J., Dapprich S., Daniels A. D., Farkas O., Foresman J., Ortiz J. V., Cioslowski J., Fox D. J., Gaussian 09, Revision A. 02, Gaussian Inc., Wallingford CT, 2009 |
| [28] | Shklover V., Nazeeruddin M. K., Grätzel M., Ovchinnikov Y. E., Appl. Organometal. Chem., 2002, 16, 635—642 |
| [29] | Fantacci S., Angelis F. D., Selloni A., J. Am. Chem. Soc., 2003, 125, 4381—4387 |
| [30] | Hagfeldt A., Grätzel M., Acc. Chem. Rev., 2000, 33, 269—277 |
| [31] | Grätzel M., Nature, 2001, 414, 338—344 |
| [1] | 何鸿锐, 夏文生, 张庆红, 万惠霖. 羟基氧化铟团簇与二氧化碳和甲烷作用的密度泛函理论研究[J]. 高等学校化学学报, 2022, 43(8): 20220196. |
| [2] | 黄汉浩, 卢湫阳, 孙明子, 黄勃龙. 石墨炔原子催化剂的崭新道路:基于自验证机器学习方法的筛选策略[J]. 高等学校化学学报, 2022, 43(5): 20220042. |
| [3] | 刘洋, 李旺昌, 张竹霞, 王芳, 杨文静, 郭臻, 崔鹏. Sc3C2@C80与[12]CPP纳米环之间非共价相互作用的理论研究[J]. 高等学校化学学报, 2022, 43(11): 20220457. |
| [4] | 周成思, 赵远进, 韩美晨, 杨霞, 刘晨光, 贺爱华. 硅烷类外给电子体对丙烯-丁烯序贯聚合的调控作用[J]. 高等学校化学学报, 2022, 43(10): 20220290. |
| [5] | 王园月, 安梭梭, 郑旭明, 赵彦英. 5-巯基-1, 3, 4-噻二唑-2-硫酮微溶剂团簇的光谱和理论计算研究[J]. 高等学校化学学报, 2022, 43(10): 20220354. |
| [6] | 程媛媛, 郗碧莹. ·OH自由基引发CH3SSC |
| [7] | 马丽娟, 高升启, 荣祎斐, 贾建峰, 武海顺. Sc, Ti, V修饰B/N掺杂单缺陷石墨烯的储氢研究[J]. 高等学校化学学报, 2021, 42(9): 2842. |
| [8] | 黄罗仪, 翁约约, 黄旭慧, 王朝杰. 车前草中黄酮类成分结构和性质的理论研究[J]. 高等学校化学学报, 2021, 42(9): 2752. |
| [9] | 钟声广, 夏文生, 张庆红, 万惠霖. 电中性团簇MCu2Ox(M=Cu2+, Ce4+, Zr4+)上甲烷和二氧化碳直接合成乙酸的理论研究[J]. 高等学校化学学报, 2021, 42(9): 2878. |
| [10] | 郑若昕, 张颖, 徐昕. 低标度XYG3双杂化密度泛函的开发与测评[J]. 高等学校化学学报, 2021, 42(7): 2210. |
| [11] | 应富鸣, 计辰儒, 苏培峰, 吴玮. 基于完全活性空间自洽场的杂化多组态密度泛函方法λ-DFCAS[J]. 高等学校化学学报, 2021, 42(7): 2218. |
| [12] | 王建, 张红星. 四配位铂磷光发射体结构与光物理性质关系的理论研究[J]. 高等学校化学学报, 2021, 42(7): 2245. |
| [13] | 胡伟, 刘小峰, 李震宇, 杨金龙. 金刚石纳米线氮空位色心的表面与尺寸效应[J]. 高等学校化学学报, 2021, 42(7): 2178. |
| [14] | 杨一莹, 朱荣秀, 张冬菊, 刘成卜. 金催化炔基苯并二𫫇英环化合成8-羟基异香豆素的理论研究[J]. 高等学校化学学报, 2021, 42(7): 2299. |
| [15] | 柳扬, 李清波, 孙杰, 赵显. Ga对在AlN衬底上直接生长石墨烯的远程催化[J]. 高等学校化学学报, 2021, 42(7): 2271. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||