非热解共价有机聚合物基氧还原电催化材料
鲍春竹, 向中华
高等学校化学学报
2023, 44 ( 5):
20220715-.
DOI:10.7503/cjcu20220715
在全球引入氢能技术助力实现碳中和目标的过程中, 高效、 低成本且长寿命的氧还原反应(ORR)阴极电催化剂具有重要作用. 近年来, 非贵金属催化剂的ORR催化活性和稳定性显著提高. 共价有机聚合物(COPs)因其可调节的孔隙率、 可修饰的骨架和周期性排列的有序结构而成为理想的分子结构定制的材料平台. 然而, 常用的高温热解合成策略中, 材料的结构变化不可预测, 真正的活性位点不明确, 阻碍了研究者对催化机理的深入探索. 非热解策略应运而生, 其可以充分发挥COP基材料可定制性的优势. 非热解COP基催化剂精确可控的结构能够为ORR催化机理的研究提供一个理想的模型, 从而指导设计催化性能更优秀的ORR电催化材料, 进一步促进材料的宏观制备. 本文从源头出发, 深入分析了ORR反应机理, 逐步归纳非热解COP基催化剂的设计原则和合成策略. 然后, 结合该领域内具有代表性的文献, 分析了非热解COP基材料电催化性能的影响因素, 系统阐述了非热解策略在ORR领域中的研究进展. 最后, 总结了本课题组对非热解COP基氧还原电催化材料的研究工作, 并进一步展望了非热解技术的发展前景及面临的挑战.
View image in article
Fig.3
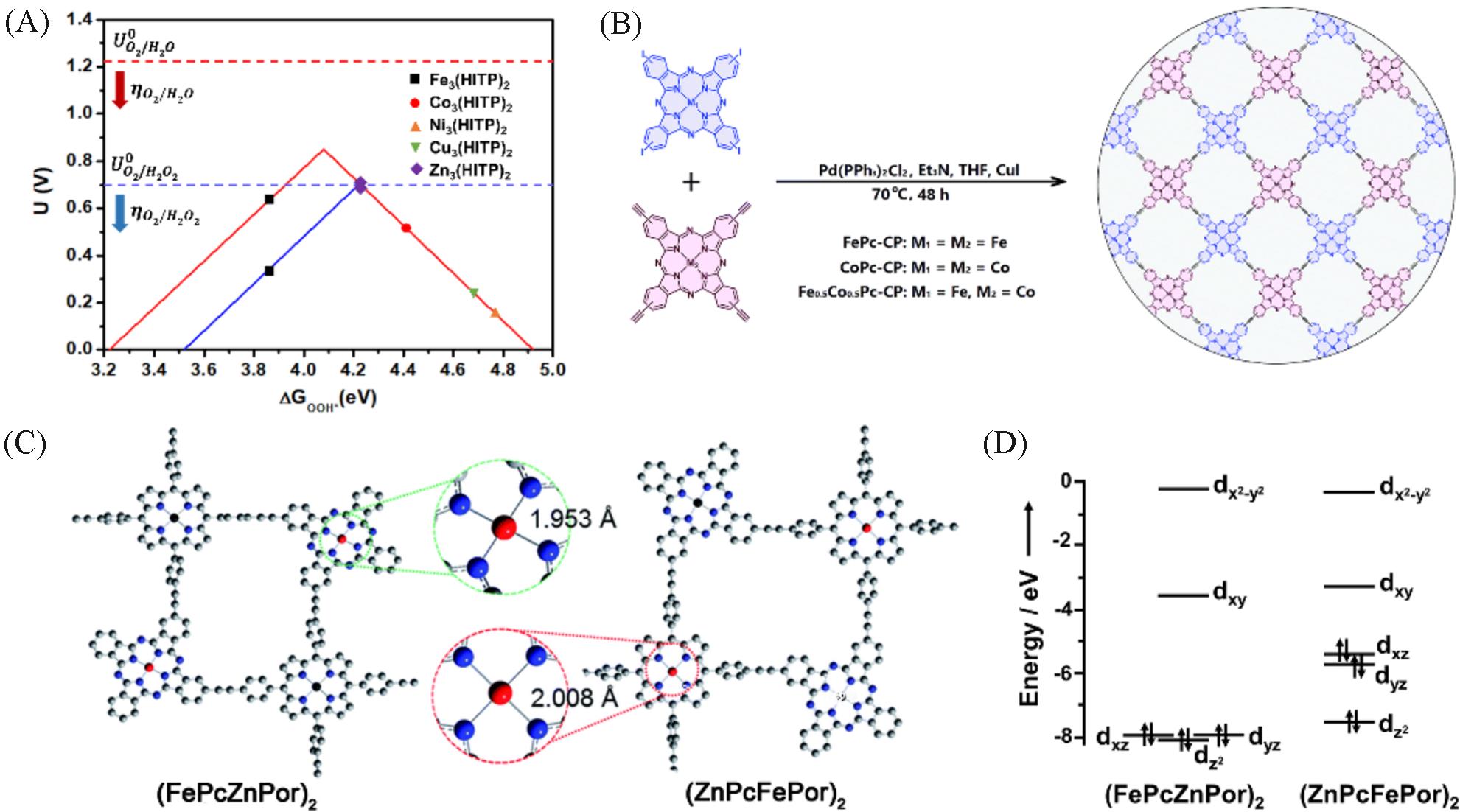
Volcano profiles of limiting potential(U) vs. ΔGOOH* for the 2e and 4e ORR processes of TM3(HITP)2 monolayers, the optimal limiting potentials indicated by the dash⁃dot lines(A)[41], schematic diagram of the synthesis(B)[49], optimized structure of (FePcZnPor)2 and (ZnPcFePor)2 moieties as the minimum repeating units for FePcZnPor⁃CMP and ZnPcFePor⁃CMP, respectively(C), schematic valence shell electronic structure of the Fe ions in (FePcZnPor)2 and (ZnPcFePor)2(D)[50]
(A) Copyright 2020, American Chemical Society; (B) Copyright 2018, the Royal Society of Chemistry; (C, D) Copyright 2018, the Royal Society of Chemistry.
正文中引用本图/表的段落
单原子催化是指金属以单原子的形式均匀单一地负载在金属、 金属氧化物、 二维材料和分子筛等载体上, 以单原子作为催化活性中心进行催化的反应. 区别于纳米或亚纳米级粒子, 单原子催化剂具有独特的量子尺寸效应、 较大的表面自由能及不饱和配位环境等优势, 能够实现活性位点的充分暴露, 最大限度地提高原子利用率. 理论上, 孤立金属位点的原子利用率可接近100%, 这对于金属资源尤其是贵金属的合理利用及成本降低影响重大. 利用非热解策略, 设计过渡金属配位中心的COP基催化材料, 其明确的周期性结构有助于制备原子级单分散的单原子催化剂. 另外, 不同过渡金属的嵌入可以调控活性中心对不同分子的吸附/脱附选择性, 从而影响反应动力学, 实现电催化性能的定向选择. 特别是Fe-N/C单原子催化剂, 其在ORR的4e反应过程中表现出优异的催化活性(O2+4H++4e→2H2O, E0=1.23 V). 这种高度均匀分散的活性位点和精确设计的几何构型为研究催化剂的构效关系提供了一个原子尺度视角. 2020年, 结合单原子过渡金属(TM=Fe, Co, Ni, Cu或Zn)和2,3,6,7,10,11-六氨基三苯官能团(HITP), Zhao等[41]通过改变TM原子来调节TM3(HITP)2单分子膜的配位环境, 改变反应中二维材料和中间产物之间的结合强度, 实现了对ORR过程选择性的定向调控. 该研究通过第一原理计算, 利用TM3(HITP)2的中间态的吉布斯自由能(|ΔGOOH*|)来评价材料的ORR性能. 绘制的极限电位火山剖面[图3(A)]中, 火山的左分支对应于HO*在催化剂上的强结合作用, 有利于H2O的形成; 而在火山的右分支中, H2O2的形成为主导过程. 在靠近火山顶峰处, H2O和H2O2的形成均有可能. 数据表明, Fe3(HITP)2倾向于4e的ORR过程, 而Cu3(HITP)2和Ni3(HITP)2中 2e的ORR过程占优势. 另外, 该课题组利用单原子催化剂独特的优势, 将TM原子优先嵌入在 TM3(HITP)2中而不是团簇中, 使得金属位点均匀分散, 最大限度地暴露其活性位点, 充分地利用其固有活性. 单原子电催化剂的可调催化活性为非热解COP基催化材料的低成本、 精准可控设计指明了方向, 也为其在清洁和可再生能源领域的应用开辟了道路.
随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种.
(A) Copyright 2020, American Chemical Society; (E) Copyright 2017, American Chemical Society. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 1 ... 随着催化机理研究的不断深入, 非热解策略制备的双金属原子催化剂也得到逐步发展[43,44]. 与单金属原子催化剂相比, 双金属原子催化剂中单原子的均匀分散和相邻双金属中心的协同作用相结合, 能够优化材料的吸附/脱附特性, 降低整体反应能垒, 实现催化活性的提高[45,46]. 另外, 双金属活性位点结构也有利于O—O键断裂, 抑制脱金属现象的发生, 有效地提高了催化剂的稳定性[47,48]. 2018年, Yao课题组[49]通过Sonogashira-Hagihara偶联反应, 制备了乙基相连的异质金属材料(Fe0.5Co0.5Pc-CPs). 由图3(B)可见, Fe和Co离子分布在异质金属材料中, 1个金属离子被相邻的第2个金属离子的4个离子紧密地包围. 与类似的单金属原子催化剂(MPc-CPs, M=Fe或Co)相比, Fe0.5Co0.5Pc-CPs在碱性介质中表现出高于MPc-CPs或商用Pt/C的ORR活性, 且密度泛函理论(DFT)计算表明双金属之间的协同作用促进了2×2e催化反应路径. Jiang课题组[50]也通过Sonogashira-Hagihara偶联反应, 制备了一系列由酞菁(Pc)和卟啉(Por)相间分散的双金属原子催化剂, 其明确的双金属催化结构为Fe-N4/C催化剂催化机理的研究提供了理想模型. 研究结果表明, 含FePc的催化剂(FePcFePor-CMP和FePcZnPor-CMP)在碱性介质中的ORR催化性能优于含FePor的催化剂(ZnPcFePor-CMP)和商用Pt/C(E1/2达到863~866 mV). 在此基础上, 该课题组进一步研究了不同聚合单体组合导致不同ORR性能的原因, 从而揭示了Fe-N4活性位点上优异ORR性能的来源.由 图3(C)可见, (FePcZnPor)2和 (ZnPcFePor)2中的Fe离子均位于四吡咯环的中心, 并与4个N原子配位. 而且FePcZnPor的Fe—N键长(1.953 nm)明显短于 ZnPcFePor(2.008 nm). Pc大环配体中较短的Fe—N键产生了比Por环更强的配位场, 导致所占dxz 轨道能量较低, 比Por更能稳定Fe2+[图3(D)]. 显然, Fe2+通常被认为是Fe-N4/C催化剂吸附O2并将O2还原为H2O或OH-的真正物种. ... 2 ... 单原子催化是指金属以单原子的形式均匀单一地负载在金属、 金属氧化物、 二维材料和分子筛等载体上, 以单原子作为催化活性中心进行催化的反应. 区别于纳米或亚纳米级粒子, 单原子催化剂具有独特的量子尺寸效应、 较大的表面自由能及不饱和配位环境等优势, 能够实现活性位点的充分暴露, 最大限度地提高原子利用率. 理论上, 孤立金属位点的原子利用率可接近100%, 这对于金属资源尤其是贵金属的合理利用及成本降低影响重大. 利用非热解策略, 设计过渡金属配位中心的COP基催化材料, 其明确的周期性结构有助于制备原子级单分散的单原子催化剂. 另外, 不同过渡金属的嵌入可以调控活性中心对不同分子的吸附/脱附选择性, 从而影响反应动力学, 实现电催化性能的定向选择. 特别是Fe-N/C单原子催化剂, 其在ORR的4e反应过程中表现出优异的催化活性(O2+4H++4e→2H2O, E0=1.23 V). 这种高度均匀分散的活性位点和精确设计的几何构型为研究催化剂的构效关系提供了一个原子尺度视角. 2020年, 结合单原子过渡金属(TM=Fe, Co, Ni, Cu或Zn)和2,3,6,7,10,11-六氨基三苯官能团(HITP), Zhao等[41]通过改变TM原子来调节TM3(HITP)2单分子膜的配位环境, 改变反应中二维材料和中间产物之间的结合强度, 实现了对ORR过程选择性的定向调控. 该研究通过第一原理计算, 利用TM3(HITP)2的中间态的吉布斯自由能(|ΔGOOH*|)来评价材料的ORR性能. 绘制的极限电位火山剖面[图3(A)]中, 火山的左分支对应于HO*在催化剂上的强结合作用, 有利于H2O的形成; 而在火山的右分支中, H2O2的形成为主导过程. 在靠近火山顶峰处, H2O和H2O2的形成均有可能. 数据表明, Fe3(HITP)2倾向于4e的ORR过程, 而Cu3(HITP)2和Ni3(HITP)2中 2e的ORR过程占优势. 另外, 该课题组利用单原子催化剂独特的优势, 将TM原子优先嵌入在 TM3(HITP)2中而不是团簇中, 使得金属位点均匀分散, 最大限度地暴露其活性位点, 充分地利用其固有活性. 单原子电催化剂的可调催化活性为非热解COP基催化材料的低成本、 精准可控设计指明了方向, 也为其在清洁和可再生能源领域的应用开辟了道路.
本文的其它图/表
-
 Fig.1
Reaction process at the three phase interface of fuel cell cathode electrode and the direction of ORR activity enhancement
Fig.1
Reaction process at the three phase interface of fuel cell cathode electrode and the direction of ORR activity enhancement
-
 Fig.2
Summary of active center design options, including single/bimetal atom(A)[41], MN4(B), MN5(C), MO4(D), MO6(E)[42], thiophene sulfide group(F) and other groups(G) engineered as active centers(A) Copyright 2020, American Chemical Society; (E) Copyright 2017, American Chemical Society.
Fig.2
Summary of active center design options, including single/bimetal atom(A)[41], MN4(B), MN5(C), MO4(D), MO6(E)[42], thiophene sulfide group(F) and other groups(G) engineered as active centers(A) Copyright 2020, American Chemical Society; (E) Copyright 2017, American Chemical Society.
-
 Fig.4
Diagram showing the electronic “push effect” of the trans axial histidine imidazole group(A)[55], proposed reaction mechanism(B)[56], optimized structures(C) and free energy diagrams(D)[59](A) Copyright 2021, Wiley-VCH; (B) Copyright 2019, Wiley-VCH; (C, D) Copyright 2020, American Chemical Society.
Fig.4
Diagram showing the electronic “push effect” of the trans axial histidine imidazole group(A)[55], proposed reaction mechanism(B)[56], optimized structures(C) and free energy diagrams(D)[59](A) Copyright 2021, Wiley-VCH; (B) Copyright 2019, Wiley-VCH; (C, D) Copyright 2020, American Chemical Society.
-
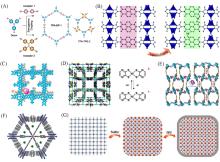 Fig.5
Summary of doping strategies, including donor/acceptor doping(A), ligand⁃n doping(B)[64], I2 doping(C—E)[36,65,66], guest doping(F)[67] and conductive polymer doping(G)[68](B) Copyright 2020, the Royal Society of Chemistry; (C) Copyright 2020, American Chemical Society; (D) Copyright 2010, American Chemical Society; (E) Copyright 2017, Wiley-VCH; (F) Copyright 2018, Springer Nature; (G) Copyright 2018, Wiley-VCH.
Fig.5
Summary of doping strategies, including donor/acceptor doping(A), ligand⁃n doping(B)[64], I2 doping(C—E)[36,65,66], guest doping(F)[67] and conductive polymer doping(G)[68](B) Copyright 2020, the Royal Society of Chemistry; (C) Copyright 2020, American Chemical Society; (D) Copyright 2010, American Chemical Society; (E) Copyright 2017, Wiley-VCH; (F) Copyright 2018, Springer Nature; (G) Copyright 2018, Wiley-VCH.
-
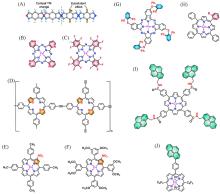 Fig.6
Design principle for substitution effects(A), construction schemes including substitution of electron⁃withdrawing groups such as pyrrole⁃N(B), F(C), thiophene⁃S(D), and NO2(E, F), electron⁃donating groups such as diphenylthiophene sulfur(G), auxiliary groups(H) and linker groups(I, J)
Fig.6
Design principle for substitution effects(A), construction schemes including substitution of electron⁃withdrawing groups such as pyrrole⁃N(B), F(C), thiophene⁃S(D), and NO2(E, F), electron⁃donating groups such as diphenylthiophene sulfur(G), auxiliary groups(H) and linker groups(I, J)
-
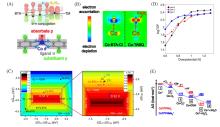 Fig.7
Schematic illustration of d⁃π conjugation in TM⁃BTA and the substituent modification strategy to regulate ORR activity of Co sites viad⁃π conjugation(A), differential charge density of *O on Co⁃BTA after the introduction of Cl and O substituents, respectively(B), the color⁃filled map of ORR activity volcano(C)[70], turnover⁃overpotential relationships for CoTPPNMe3+ at different pH values(D), and calculated relative free energy changes for ORR(E)[73](A—C) Copyright 2021, Wiley-VCH; (D, E) Copyright 2020, American Chemical Society.
Fig.7
Schematic illustration of d⁃π conjugation in TM⁃BTA and the substituent modification strategy to regulate ORR activity of Co sites viad⁃π conjugation(A), differential charge density of *O on Co⁃BTA after the introduction of Cl and O substituents, respectively(B), the color⁃filled map of ORR activity volcano(C)[70], turnover⁃overpotential relationships for CoTPPNMe3+ at different pH values(D), and calculated relative free energy changes for ORR(E)[73](A—C) Copyright 2021, Wiley-VCH; (D, E) Copyright 2020, American Chemical Society.
-
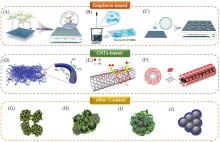 Fig.8
The published supported type with pyrolysis⁃free mainly classified into graphene⁃based(A—C)[37,84,85], CNTs⁃based(D—F)[86—88] and other C⁃based(G—J)[89—91] materials(A) Copyright 2018, Wiley-VCH; (B) Copyright 2019, the Royal Society of Chemistry; (C) Copyright 2019, American Association for the Advancement of Science; (D) Copyright 2020, Elsevier; (E) Copyright 2015, Elsevier; (F) Copyright 2014, American Chemical Society; (G, H) Copyright 2014, Elsevier; (I) Copyright 2017, Springer Nature; (J) Copyright 2015, the Royal Society of Chemistry.
Fig.8
The published supported type with pyrolysis⁃free mainly classified into graphene⁃based(A—C)[37,84,85], CNTs⁃based(D—F)[86—88] and other C⁃based(G—J)[89—91] materials(A) Copyright 2018, Wiley-VCH; (B) Copyright 2019, the Royal Society of Chemistry; (C) Copyright 2019, American Association for the Advancement of Science; (D) Copyright 2020, Elsevier; (E) Copyright 2015, Elsevier; (F) Copyright 2014, American Chemical Society; (G, H) Copyright 2014, Elsevier; (I) Copyright 2017, Springer Nature; (J) Copyright 2015, the Royal Society of Chemistry.
-
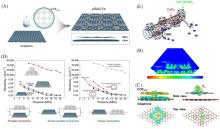 Fig.9
Synthesis route of the pfSAC⁃Fe catalyst(A), the electron localization function of pfSAC⁃Fe(B), differential charge density distribution on pfSAC⁃Fe before and after the absorption of oxygen(C), electronic conductivity studies(D)[37] and schematic structure of UiO⁃66⁃NO2@CoCNT(E)[93](A—D) Copyright 2019, American Association for the Advancement of Science; (E) Copyright 2019, American Chemical Society.
Fig.9
Synthesis route of the pfSAC⁃Fe catalyst(A), the electron localization function of pfSAC⁃Fe(B), differential charge density distribution on pfSAC⁃Fe before and after the absorption of oxygen(C), electronic conductivity studies(D)[37] and schematic structure of UiO⁃66⁃NO2@CoCNT(E)[93](A—D) Copyright 2019, American Association for the Advancement of Science; (E) Copyright 2019, American Chemical Society.
-
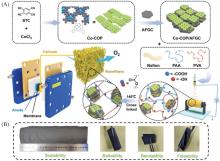 Fig.10
Schematic illustration of the preparation process for the cross⁃linked nanofiber electrode(A) and photos of cross⁃linked nanofiber membrane obtained by electrospinning(B)[94]Copyright 2022, Wiley-VCH.
Fig.10
Schematic illustration of the preparation process for the cross⁃linked nanofiber electrode(A) and photos of cross⁃linked nanofiber membrane obtained by electrospinning(B)[94]Copyright 2022, Wiley-VCH.
-
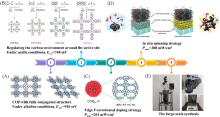 Fig.11
Summary of representative references about pyrolysis⁃free strategies in our group, including the modulation of intrinsic properties(A[92], B[96], C[98]), application of devices(D)[94] and large⁃scale preparation(E)[99](A) Copyright 2019, American Chemical Society; (B) Copyright 2022, Springer Nature; (C) Copyright 2022, Wiley-VCH; (D) Copyright 2022, Wiley-VCH; (E) Copyright 2022, Wiley-VCH.
Fig.11
Summary of representative references about pyrolysis⁃free strategies in our group, including the modulation of intrinsic properties(A[92], B[96], C[98]), application of devices(D)[94] and large⁃scale preparation(E)[99](A) Copyright 2019, American Chemical Society; (B) Copyright 2022, Springer Nature; (C) Copyright 2022, Wiley-VCH; (D) Copyright 2022, Wiley-VCH; (E) Copyright 2022, Wiley-VCH.
|


{kind=link}