高等学校化学学报
2023, 44 (
):
20220715-.
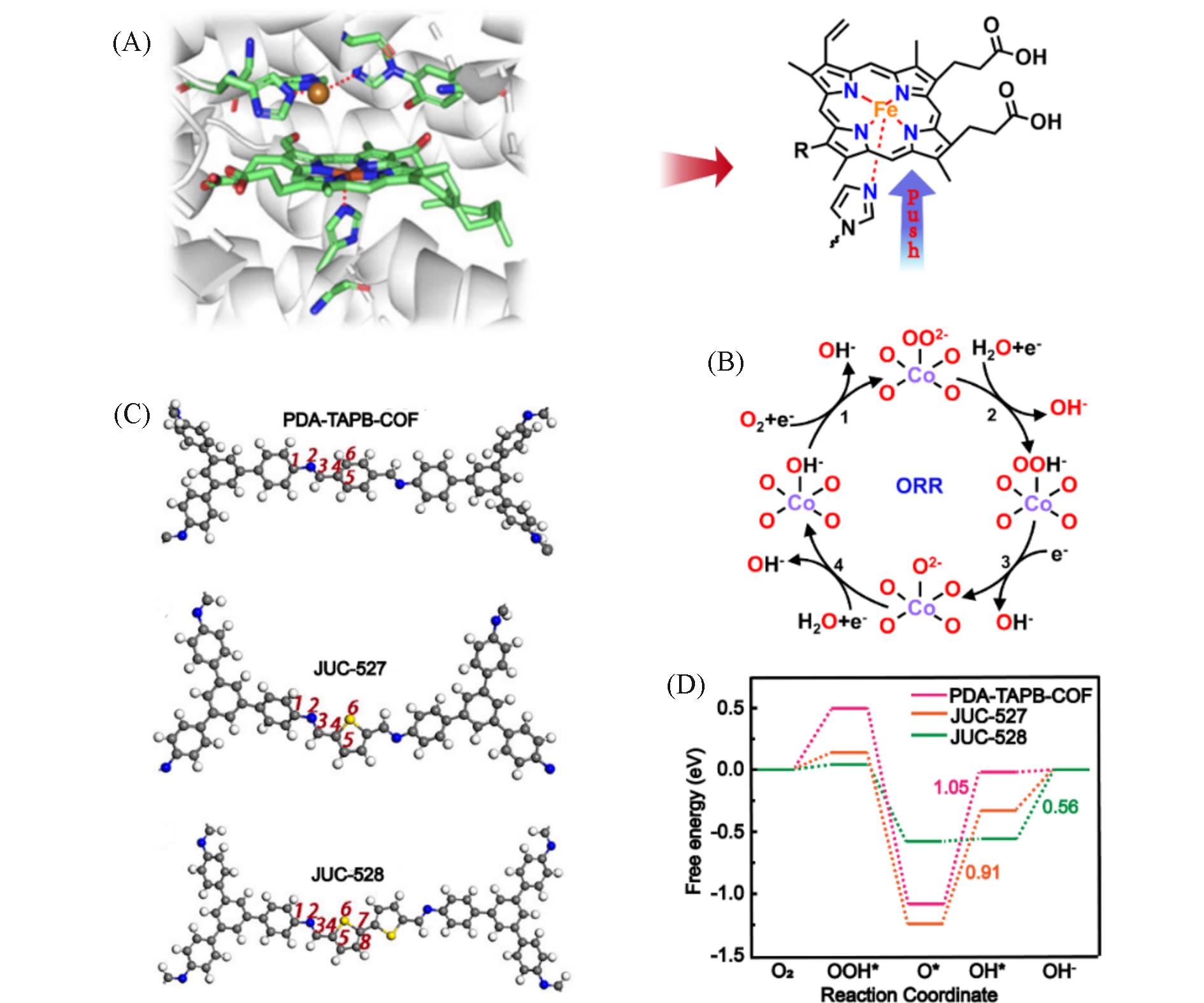
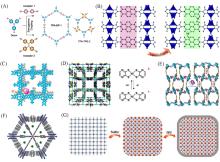
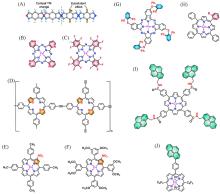
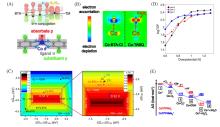
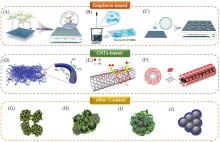
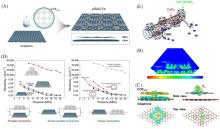
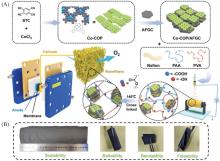
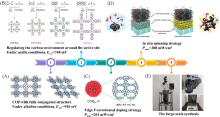
在全球引入氢能技术助力实现碳中和目标的过程中, 高效、 低成本且长寿命的氧还原反应(ORR)阴极电催化剂具有重要作用. 近年来, 非贵金属催化剂的ORR催化活性和稳定性显著提高. 共价有机聚合物(COPs)因其可调节的孔隙率、 可修饰的骨架和周期性排列的有序结构而成为理想的分子结构定制的材料平台. 然而, 常用的高温热解合成策略中, 材料的结构变化不可预测, 真正的活性位点不明确, 阻碍了研究者对催化机理的深入探索. 非热解策略应运而生, 其可以充分发挥COP基材料可定制性的优势. 非热解COP基催化剂精确可控的结构能够为ORR催化机理的研究提供一个理想的模型, 从而指导设计催化性能更优秀的ORR电催化材料, 进一步促进材料的宏观制备. 本文从源头出发, 深入分析了ORR反应机理, 逐步归纳非热解COP基催化剂的设计原则和合成策略. 然后, 结合该领域内具有代表性的文献, 分析了非热解COP基材料电催化性能的影响因素, 系统阐述了非热解策略在ORR领域中的研究进展. 最后, 总结了本课题组对非热解COP基氧还原电催化材料的研究工作, 并进一步展望了非热解技术的发展前景及面临的挑战.


{kind=link}