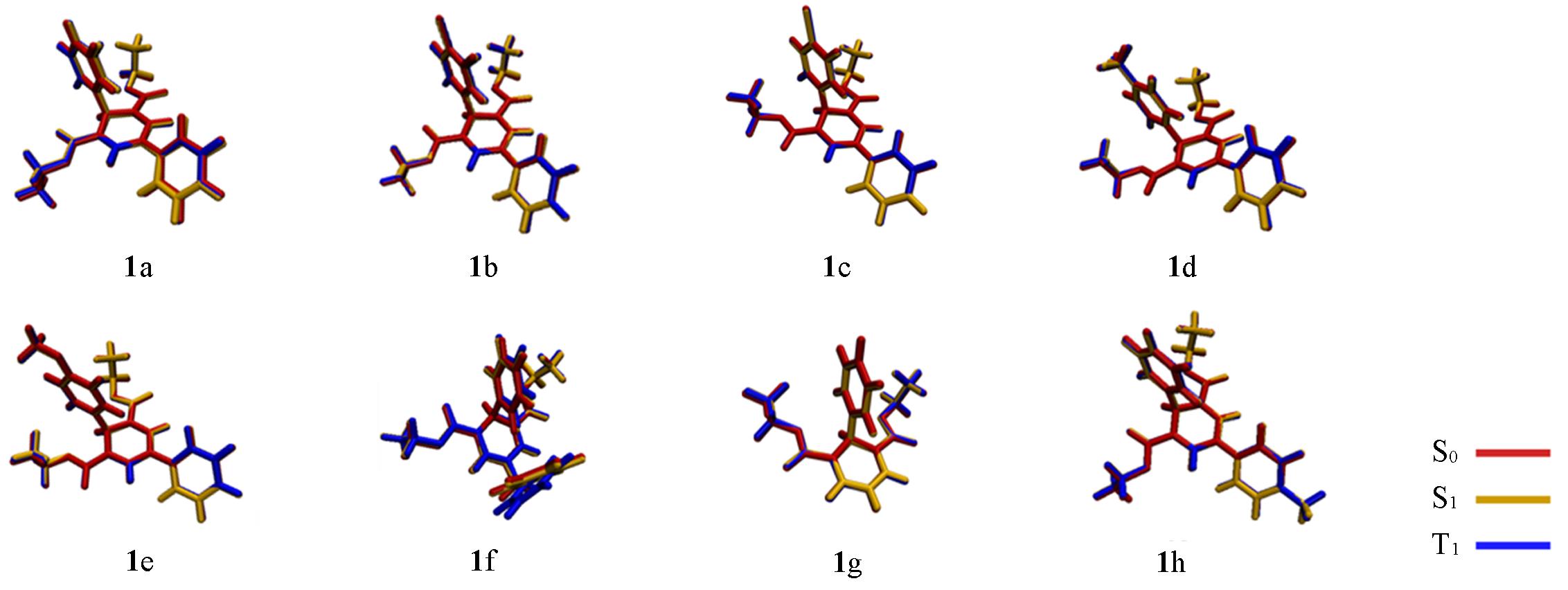
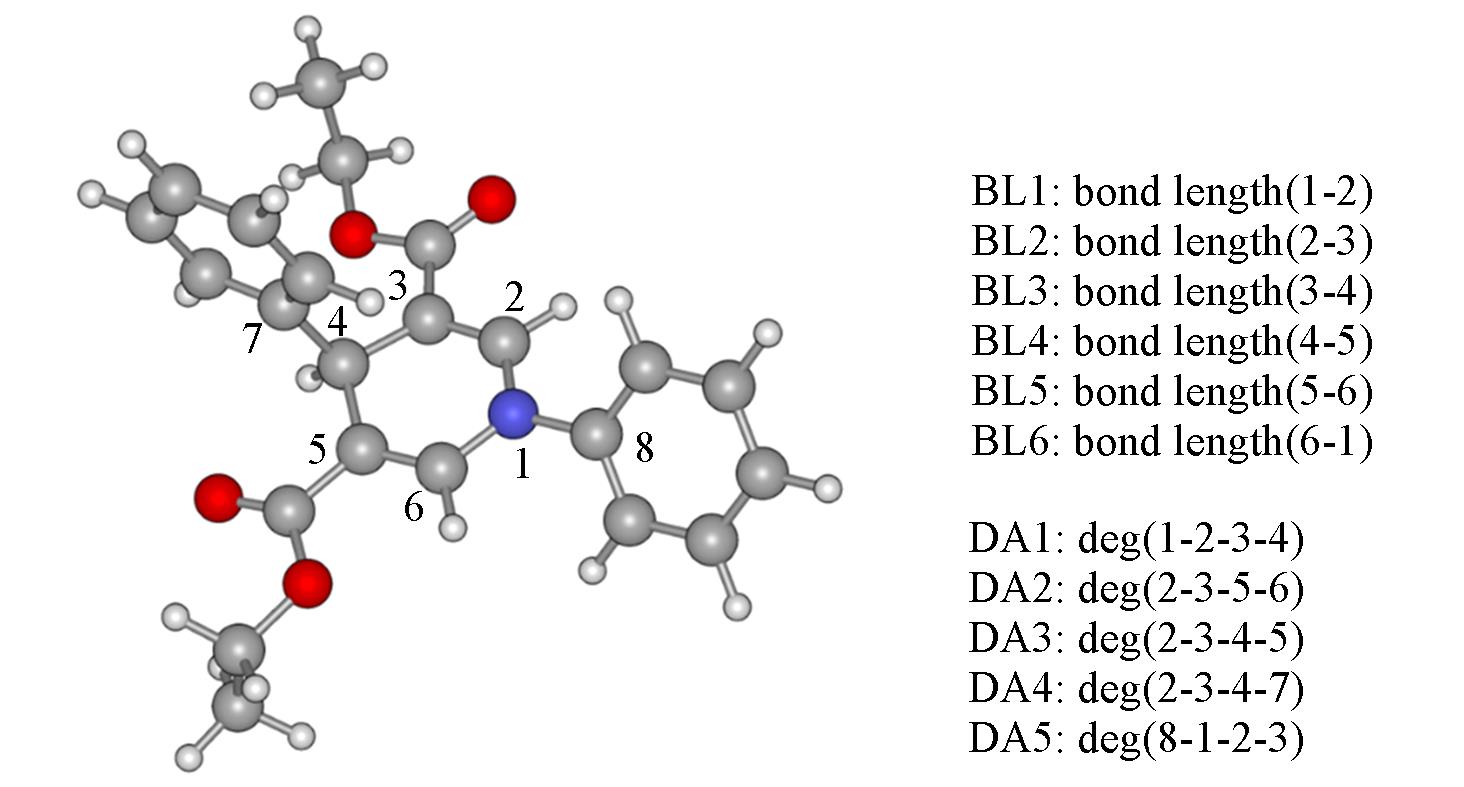
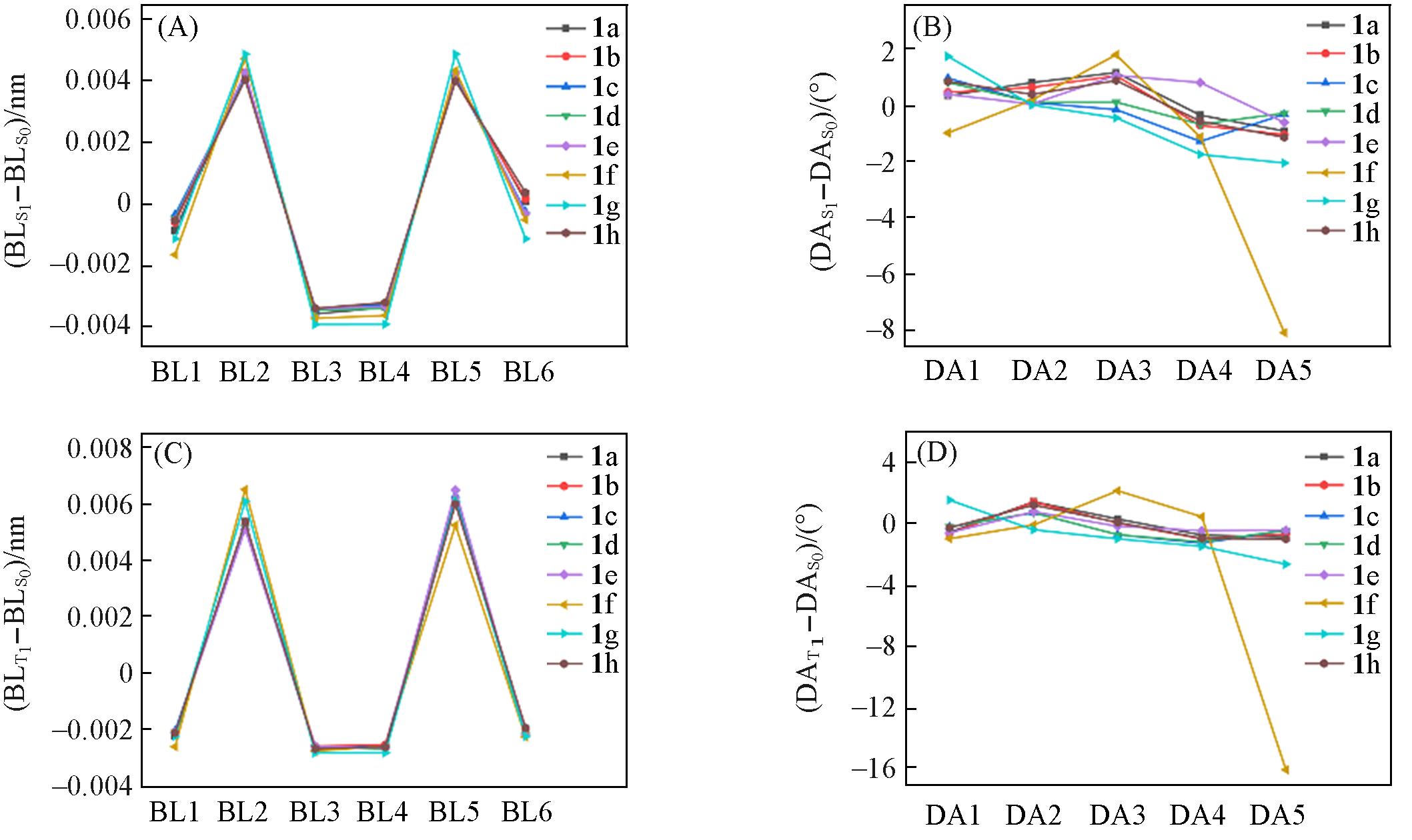
| [1] |
Wang C., Lu Z., Org. Lett., 2017, 19(21), 5888—5891
|
| [2] |
Zhang Y., Tanabe Y., Kuriyama S., Nishibayashi Y., J. Org. Chem., 2021, 86(18), 12577—12590
|
| [3] |
Zhong Q., Fan Q., Yan H., Tetrahedron Lett., 2017, 58(13), 1292—1295
|
| [4] |
Yan Z., Li J., Zheng Y., Miao Z., Chin. J. Chem. Educ., 2023, 44(18), 1—16
|
| [5] |
Chaudhari S. R., Shirkhedkar A. A., Chin. J. Org. Chem., 2021, 51(3), 268—277
|
| [6] |
Liu Y., Tan H., Yan H., Song X., Chem. Biol. Drug Des., 2013, 82(5), 567—578
|
| [7] |
Manna D., Bhuyan R., Ghosh R., J. Mol. Model., 2018, 24(12), 340
|
| [8] |
Ong S. T., Nam Y. W., Nasburg J. A., Ramanishka A., Ng X. R., Zhuang Z., Goay S. S. M., Nguyen H. M., Singh L., Singh V., Rivera A., Eyster M. E., Xu Y., Alper S. L., Wulff H., Zhang M., Chandy K. G., PNAS, 2025, 122(18), e2425494122
|
| [9] |
Trivedi A. R., Dodiya D. K., Dholariya B. H., Kataria V. B., Bhuva V. R., Shah V. H., Bioorg. Med. Chem. Lett., 2011, 21(18), 5181—5183
|
| [10] |
Valente S., Mellini P., Spallotta F., Carafa V., Nebbioso A., Polletta L., Carnevale I., Saladini S., Trisciuoglio D., Gabellini C., Tardugno M., Zwergel C., Cencioni C., Atlante S., Moniot S., Steegborn C., Budriesi R., Tafani M., Del Bufalo D., Altucci L., Gaetano C., Mai A., J. Med. Chem., 2016, 59(4), 1471—1491
|
| [11] |
Zhu H., Chen S. J., Lin Y. X., Zhong Q. D., Sun W. J., Zhang X. J., Z. Kristallogr. New Cryst. Struct., 2019, 234(4), 793—794
|
| [12] |
Chen X., Niu N., Li D., Zhang Z., Zhuang Z., Yan D., Li J., Zhao Z., Wang D., Tang B. Z., Adv. Funct. Mater., 2023, 33(8), 2211571
|
| [13] |
Lian C., Zhang J., Mo F., Org. Chem. Front., 2024, 11(4), 1140—1149
|
| [14] |
Fan Q., Tan H., Li P., Yan H., New J. Chem., 2018, 42(20), 16795—16805
|
| [15] |
Zhu X., Li W., Yan H., Zhong R., J. Photochem. Photobiol. A: Chem., 2012, 241, 13—20
|
| [16] |
Zhu X. H., Ni C. L., Song X. Q., Yan H., Zhong R. G., Chin. J. Org. Chem., 2010, 30(2), 276—281
|
| [17] |
Sun W., Fan Q., Yan H., J. Photochem. Photobiol. A: Chem., 2018, 359, 33—39
|
| [18] |
Tan H. B., Zhao Z. C., Ma Z. S., Yan H., Tetrahedron, 2018, 74(5), 529—534
|
| [19] |
Wang S., Wang Y., Ge C., Sun R., Wang H., Yan H., J. Mol. Struct., 2023, 1273, 134316
|
| [20] |
Zhong Q. D., Study on Photochemical Reactions and Mechanism of 1,4⁃Dihydropyridine Derivatives, Beijing University of Technology, Beijing, 2017
|
|
钟启迪. 1,4-二氢吡啶衍生物的光化学反应及反应机理研究, 北京: 北京工业大学, 2017
|
| [21] |
Sun R., Song X., Wang S., Zhang X., Yan H., Wang Y., Chin. Chem. Lett., 2023, 34(12), 108183
|
| [22] |
Zhang X., Wei C., Zhang Y., Yan H., Li P., J. Mol. Struct., 2024, 1306, 137893
|
| [23] |
Frisch M. J., Pople J. A., Binkley J. S., J. Chem. Phys., 1984, 80(7), 3265—3269
|
| [24] |
Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem., 2011, 32(7), 1456—1465
|
| [25] |
Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem., 1994, 98(45), 11623—11627
|
| [26] |
Cammi R., Tomasi J., J. Chem. Phys., 1994, 100(10), 7495—7502
|
| [27] |
Miertuš S., Scrocco E., Tomasi J., Chem. Phys., 1981, 55(1), 117—129
|
| [28] |
Lu T., Chen F., J. Comput. Chem., 2012, 33(5), 580—592
|
| [29] |
Lu T., J. Chem. Phys., 2024, 161(8), 082503
|
| [30] |
Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 1996, 14(1), 33—38
|
| [31] |
Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams Y., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J., Gaussian 16, Rev. B.01, Wallingford CT, Gaussian Inc., 2016
|
| [32] |
Fan Q., Li P., Yan H., J. Photochem. Photobiol. A: Chem., 2018, 358, 51—60
|
| [33] |
He Y., Wang H., Ge C., Yan H., J. Mol. Struct., 2023, 1281, 135167
|
| [34] |
Li P., Wang S., Tian N., Yan H., Wang J., Song X., Org. Biomol. Chem., 2021, 19(17), 3882—3892
|
| [35] |
Costa R. A., Pitt P. O., Pinheiro M. L. B., Oliveira K. M. T., Salomé K. S., Barison A., Costa E. V., Spectrochim. Acta A: Mol. Biomol. Spectrosc., 2017, 174, 94—104
|
 ), 孙国辉1
), 孙国辉1