

高等学校化学学报 ›› 2017, Vol. 38 ›› Issue (9): 1611.doi: 10.7503/cjcu20170189
钱梦丹, 罗伟, 倪哲明( ), 夏盛杰, 薛继龙, 蒋军辉
), 夏盛杰, 薛继龙, 蒋军辉
收稿日期:2017-03-29
出版日期:2017-09-10
发布日期:2017-08-25
作者简介:联系人简介: 倪哲明, 女, 博士, 教授, 博士生导师, 主要从事计算化学及纳米无机光催化材料研究. E-mail:基金资助:
QIAN Mengdan, LUO Wei, NI Zheming*(), XIA Shengjie, XUE Jilong, JIANG Junhui
Received:2017-03-29
Online:2017-09-10
Published:2017-08-25
Contact:
NI Zheming
E-mail:jchx@zjut.edu.cn
Supported by:摘要:
采用密度泛函理论研究了Pd(111)面和Ru-Pd(111)面的性质及对糠醛的吸附. 原子尺寸因素、 相对键长、 形成能及d带中心等计算结果表明, Ru-Pd(111)面比Pd(111)面稳定且活性强, Ru的修饰优化了Pd(111)面的几何构型. 糠醛在Pd(111)面及Ru-Pd(111)面的初始吸附位分别为P(top-bridge)位及P(Pd-fcc-Ru-fcc)位时, 吸附能最大, 吸附构型最稳定. 由电荷布局和差分电荷密度可得, 糠醛在 Ru-Pd(111)面上电荷转移数更多, 相互作用更强烈, 因此吸附能更大. 分析态密度可知, 产生吸附的主要原因是位于-7.34 eV处至费米能级处的p, d轨道杂化. 吸附于Ru-Pd(111)面后糠醛分子的p轨道向低能级偏移程度更明显, 使Ru改性后的Pd催化剂具有更好的催化活性.
TrendMD:
钱梦丹, 罗伟, 倪哲明, 夏盛杰, 薛继龙, 蒋军辉. Ru修饰前后Pd(111)面的性质及对糠醛吸附的比较研究. 高等学校化学学报, 2017, 38(9): 1611.
QIAN Mengdan, LUO Wei, NI Zheming, XIA Shengjie, XUE Jilong, JIANG Junhui. Comparative Study on the Properties and Adsorption of Furfural of Pd(111) Surface Before and After Ru Modification†. Chem. J. Chinese Universities, 2017, 38(9): 1611.
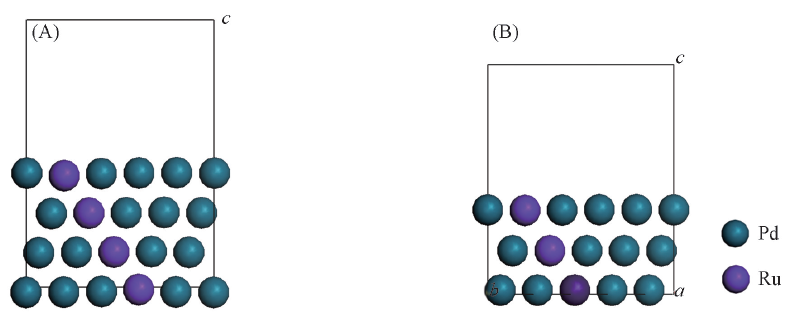
Fig.1 Structure of Ru-Pd(111)[N=4(A), 3(B)] plane surfaces model
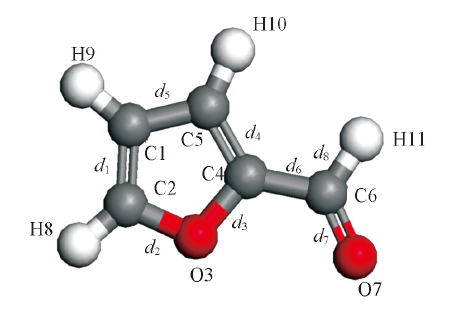
Fig.3 Structure of furfural
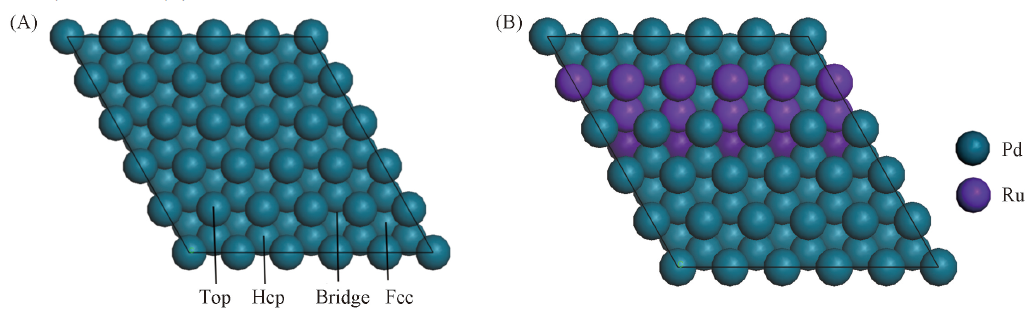
Fig.4 Structure of Pd(111)(A) and Ru-Pd(111)(B) plane surface model
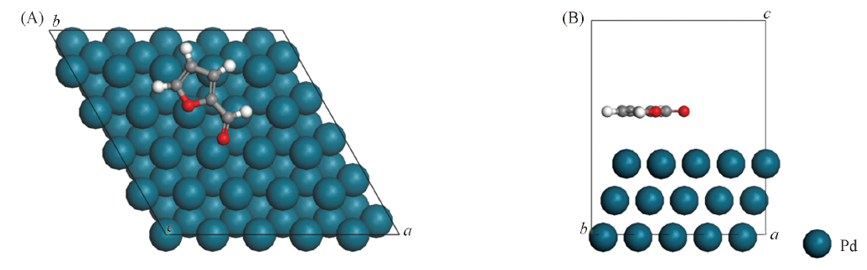
Fig.5 Top(A) and side(B) views of furfural molecule after adsorption on Pd(111) surface
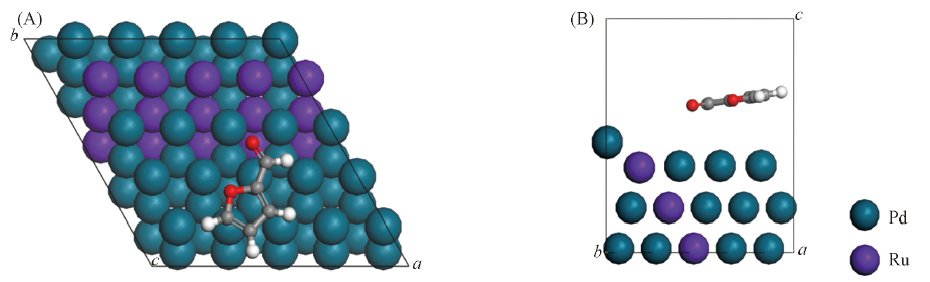
Fig.6 Top(A) and side(B) views of furfural molecule after adsorption on Ru-Pd(111) surface
| Adsorption site (Pd-O3-Pd-O7) | Eads/(kJ·mol-1) | Adsorption site (Pd-O3-Pd-O7) | Eads/(kJ·mol-1) | ||
|---|---|---|---|---|---|
| Pd(111) | Ru-Pd(111) | Pd(111) | Ru-Pd(111) | ||
| Top-top | -77.86 | -88.23 | Fcc-top | -77.48 | -110.58 |
| Top-hcp | -77.58 | -88.07 | Fcc-hcp | -76.30 | -86.63 |
| Top-fcc | -77.59 | -85.25 | Fcc-fcc | -72.50 | -85.62 |
| Top-bridge | -79.44 | -89.02 | Fcc-bridge | -75.70 | -86.25 |
| Hcp-top | -75.14 | -140.01 | Bridge-top | -71.20 | -138.32 |
| Hcp-hcp | -75.24 | -118.98 | Bridge-hcp | -75.39 | -118.78 |
| Hcp-fcc | -76.61 | -87.35 | Bridge-fcc | -77.34 | -81.26 |
| Hcp-bridge | -75.20 | -138.32 | Bridge-bridge | -75.84 | -134.08 |
Table 1 Adsorption energy(Eads) of furfural molecule on Pd(111) and Ru-Pd(111) surfaces
| Adsorption site (Pd-O3-Pd-O7) | Eads/(kJ·mol-1) | Adsorption site (Pd-O3-Pd-O7) | Eads/(kJ·mol-1) | ||
|---|---|---|---|---|---|
| Pd(111) | Ru-Pd(111) | Pd(111) | Ru-Pd(111) | ||
| Top-top | -77.86 | -88.23 | Fcc-top | -77.48 | -110.58 |
| Top-hcp | -77.58 | -88.07 | Fcc-hcp | -76.30 | -86.63 |
| Top-fcc | -77.59 | -85.25 | Fcc-fcc | -72.50 | -85.62 |
| Top-bridge | -79.44 | -89.02 | Fcc-bridge | -75.70 | -86.25 |
| Hcp-top | -75.14 | -140.01 | Bridge-top | -71.20 | -138.32 |
| Hcp-hcp | -75.24 | -118.98 | Bridge-hcp | -75.39 | -118.78 |
| Hcp-fcc | -76.61 | -87.35 | Bridge-fcc | -77.34 | -81.26 |
| Hcp-bridge | -75.20 | -138.32 | Bridge-bridge | -75.84 | -134.08 |
| Model | d1/nm | d2/nm | d3/nm | d4/nm | d5/nm | d6/nm | d7/nm | d8/nm |
|---|---|---|---|---|---|---|---|---|
| Free-furfural | 0.1399 | 0.1382 | 0.1402 | 0.1414 | 0.1420 | 0.1423 | 0.1274 | 0.1112 |
| P(top-bridge)/Pd(111) | 0.1392 | 0.1378 | 0.1400 | 0.1409 | 0.1417 | 0.1427 | 0.1259 | 0.1110 |
| P(Pd-fcc-Ru-fcc)/Ru-Pd(111) | 0.1400 | 0.1378 | 0.1401 | 0.1413 | 0.1419 | 0.1426 | 0.1271 | 0.1110 |
| Δd1(%) | -0.50 | -0.29 | -0.14 | -0.35 | -0.21 | 0.28 | -1.18 | -0.18 |
| Δd2(%) | 0.07 | -0.29 | -0.07 | -0.07 | -0.07 | 0.21 | -0.24 | -0.18 |
Table 2 Structure parameters of the most stable adsorption configuration of furfural on Pd(111) and Ru-Pd(111) surfaces*
| Model | d1/nm | d2/nm | d3/nm | d4/nm | d5/nm | d6/nm | d7/nm | d8/nm |
|---|---|---|---|---|---|---|---|---|
| Free-furfural | 0.1399 | 0.1382 | 0.1402 | 0.1414 | 0.1420 | 0.1423 | 0.1274 | 0.1112 |
| P(top-bridge)/Pd(111) | 0.1392 | 0.1378 | 0.1400 | 0.1409 | 0.1417 | 0.1427 | 0.1259 | 0.1110 |
| P(Pd-fcc-Ru-fcc)/Ru-Pd(111) | 0.1400 | 0.1378 | 0.1401 | 0.1413 | 0.1419 | 0.1426 | 0.1271 | 0.1110 |
| Δd1(%) | -0.50 | -0.29 | -0.14 | -0.35 | -0.21 | 0.28 | -1.18 | -0.18 |
| Δd2(%) | 0.07 | -0.29 | -0.07 | -0.07 | -0.07 | 0.21 | -0.24 | -0.18 |
| Atom | Charge/e | ||
|---|---|---|---|
| Free furfural | Furfural/Pd(111) | Furfural/Ru-Pd(111) | |
| C1 | -0.059 | -0.097 | -0.102 |
| C2 | 0.196 | 0.153 | 0.186 |
| O3 | -0.371 | -0.327 | -0.346 |
| C4 | 0.328 | 0.272 | 0.263 |
| C5 | -0.103 | -0.118 | -0.092 |
| C6 | 0.169 | 0.134 | 0.170 |
| O7 | -0.351 | -0.318 | -0.315 |
| H8 | 0.050 | 0.099 | 0.091 |
| H9 | 0.062 | 0.100 | 0.093 |
| H10 | 0.048 | 0.096 | 0.086 |
| H11 | 0.031 | 0.064 | 0.057 |
| Tol | 0 | 0.058 | 0.091 |
Table 3 Mulliken atomic charge populations of furfural molecule at advantage adsorption site on the Pd(111) and Ru-Pd(111) surfaces
| Atom | Charge/e | ||
|---|---|---|---|
| Free furfural | Furfural/Pd(111) | Furfural/Ru-Pd(111) | |
| C1 | -0.059 | -0.097 | -0.102 |
| C2 | 0.196 | 0.153 | 0.186 |
| O3 | -0.371 | -0.327 | -0.346 |
| C4 | 0.328 | 0.272 | 0.263 |
| C5 | -0.103 | -0.118 | -0.092 |
| C6 | 0.169 | 0.134 | 0.170 |
| O7 | -0.351 | -0.318 | -0.315 |
| H8 | 0.050 | 0.099 | 0.091 |
| H9 | 0.062 | 0.100 | 0.093 |
| H10 | 0.048 | 0.096 | 0.086 |
| H11 | 0.031 | 0.064 | 0.057 |
| Tol | 0 | 0.058 | 0.091 |
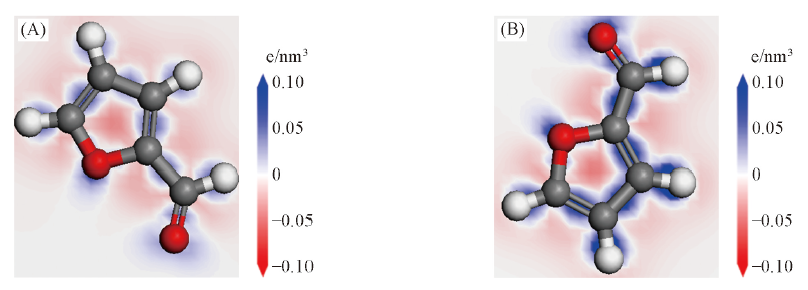
Fig.7 Deformation density of furfural adsorption on Pd(111)(A) and Ru-Pd(111)(B) surfaces
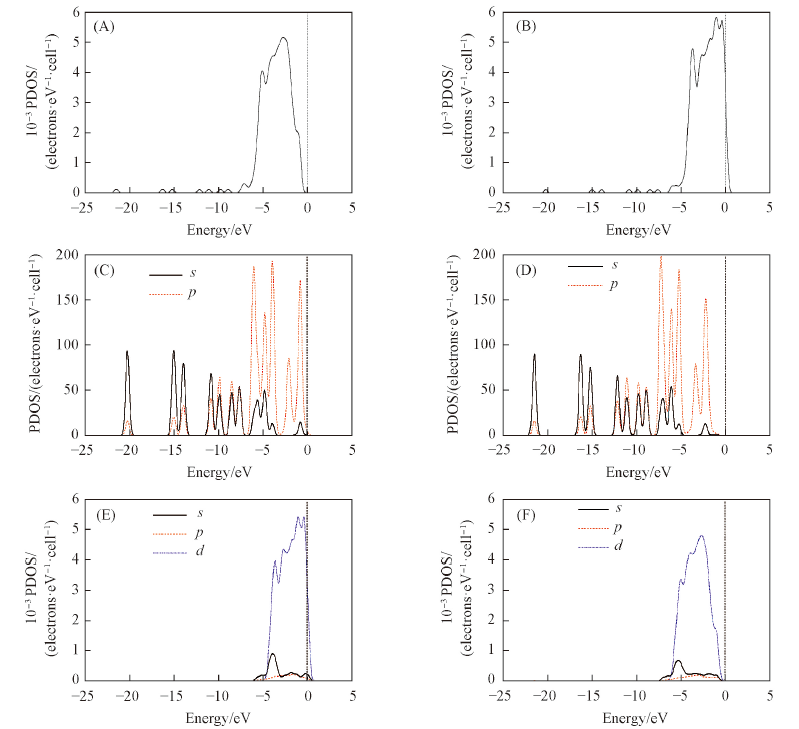
Fig.8 Densities of states of furfural adsorption on Pd(111) and Ru-Pd(111) surfaces(A) Total DOS of furfural and Pd(111) surface after adsorption; (B) total DOS of furfural and Ru-Pd(111) surface after adsorption; (C) s,p orbitals of furfual molecule on Pd(111) after adsorption; (D) s,p orbitals of furfual molecule on Ru-Pd(111) after adsorption; (E) s,p,d orbitals of Pd(111) surface after adsorption; (F) s,p,d orbitals of Ru-Pd(111) surface after adsorption.
| [1] | Juben N. C., George W. H., James A. D., Angew. Chem., 2007, 119, 7298—7318 |
| [2] | George W. H., Sara I., Avelino C., Chem. Rev., 2006, 106, 4044—4098 |
| [3] | Joseph J. B., Gene R. P., Green Chem., 2010, 12, 539—554 |
| [4] | Amanda-Lynn M., Peter J. A., Chem. Eur. J., 2010, 16, 4970—4980 |
| [5] | Saqib S. T., Lasse R., Andress R., Energy,2011, 36, 2328—2342 |
| [6] | Tao R. R., Song H. L., Chou L. J., J. Mol. Catal. A-Chem., 2012, 357, 11—18 |
| [7] | Pierre G., Chem. Soc. Rev., 2012, 41, 1538—1558 |
| [8] | Saikat D., Sudipta D., Basudeb S., Md I.A., Catal. Sci. Technol., 2012, 2, 2025—2036 |
| [9] | Yu Y. X., Zhang X. H., Wang T. J., Xu Y., Ma L. L., Zhang Q., Zhang L. M., Chem. J. Chinese Universities, 2013, 34(9), 2178—2183 |
| (于玉肖, 张兴华, 王铁军, 徐莹, 马隆龙, 张琦, 张丽敏.高等学校化学学报,2013, 34(9), 2178—2183) | |
| [10] | Wu J., Gao G., Li J. L., Sun P., Long X. D., Li F. W., Appl. Catal. B Environ., 2017, 203, 227—236 |
| [11] | Milan H., Katarina F., Catal. Commun., 2012, 24, 100—104 |
| [12] | Zhang C., Lai Q. H., Joseph H. H., Catal. Commun., 2017, 89, 77—80 |
| [13] | Rajesh V. S., Umashankar D., Ramaswami S., Ajay K. D., Appl. Catal. A Gen., 2013, 454, 127—136 |
| [14] | Bhogeswararao S., Srinivas D., J. Catal., 2011, 327, 65—77 |
| [15] | Zhao Y. Y., Environ Chem. Lett., 2014, 12, 185—190 |
| [16] | Surapas S. D. E. R., Catal Lett., 2011, 141, 784—791 |
| [17] | Zheng R. Y., Zhu Y. X., Chen J. G., Chem. Cat. Chem., 2011, 3, 578—581 |
| [18] | Zheng R. Y., Humbert M. P., Zhu Y. X., Chen J. G., Catal. Sci. Technol., 2011, 1, 638—643 |
| [19] | Sitthisa S., Pham T., Prasomsri T., Sooknoi T., Mallinson R. G., Resasco D. E., J. Catal., 2011, 280, 17—27 |
| [20] | Sitthisa S., An W., Resasco D. E., J. Catal., 2011, 284, 90—101 |
| [21] | Qiu J. S., Zhang H. Z., Wang X. N., Han H. M., Liang C. H., Li C., React. Kinet. Catal. Lett., 2006, 88, 269—275 |
| [22] | Obaid F. A., Sarwat L., Peter J. M., Germma L. B., Daniel R. J., Xi L., Jenniffr K. E., David J. M., David K. K., Graham J. H., Catal. Sci. Technol., 2016, 6, 234—242 |
| [23] | Sun X. L., Huo R. P., Bu Y. X., Li J. L., Chem. J. Chinese Universities, 2015, 36(8), 1570—1575 |
| (孙小丽, 霍瑞萍, 步宇翔, 李吉来.高等学校化学学报,2015, 36(8), 1570—1575) | |
| [24] | Liu J. H., Lu C. Q., Jin. C., Guo Y., Wang G. C., Chem. Res. Chinese Universities, 2016, 32(2), 234—241 |
| [25] | Yu Y. X., Phys. Chem. Chem. Phys., 2013, 15, 16819—16827 |
| [26] | Gholizadeh R., Yu Y. X., Appl. Surf. Sci., 2015, 357, 1187—1195 |
| [27] | Chen R. F., Xia W. S., Wan H. L., Chem. J. Chinese Universities, 2015, 36(9), 1743—1751 |
| (陈蓉芳, 夏文生, 万惠霖.高等学校化学学报,2015, 36(9), 1743—1751) | |
| [28] | Jiang J. H., Xia S. J., Ni Z. M., Zhang L. Y., Chem. J. Chinese Universities, 2016, 37(4), 693—700 |
| (蒋军辉, 夏盛杰, 倪哲明, 张连阳.高等学校化学学报,2016, 37(5), 693—700) | |
| [29] | Cao Y. Y., Jiang J. H., Ni Z. M., Xia S. J., Qian M. D., Xue J. L., Chem. J. Chinese Universities, 2016, 37(7), 1342—1350 |
| (曹勇勇, 蒋军辉, 倪哲明, 夏盛杰, 钱梦丹, 薛继龙.高等学校化学学报,2016, 37(7), 1342—1350) | |
| [30] | Ge Q., Jenkins S. J., King D. A., Chem. Phys. Lett., 2000, 327, 125—130 |
| [31] | Perdew J. P., Chewary J. A., Vosko S. H., Jackson K. A., Pedersone M. R., Singh D. J., Fiolhais C., Phys. Rev. B, 1992, 46, 6671—6687 |
| [32] | Tao J., Yao Z.J., Xue F., Fundamentals of Material Science, Chemical Industry Press, Beijing, 2006, 50—51 |
| (陶杰, 姚正军, 薛烽. 材料科学基础, 北京: 化学工业出版社, 2006, 50—51) | |
| [33] | Palotas K., Bako I., Bugyi L., Appl. Surf. Sci., 2016, 389, 1094—1103 |
| [34] | Vorotnikov V., Mpourmpakis G., Vlachos D. G., ACS. Catal., 2012, 2, 2496—2504 |
| [35] | Liu R. Q., Comput. Theor. Chem., 2013, 1019, 141—145 |
| [36] | Teng B. T., Zhao Y., Wu F. M., Wen X. D., Chen Q. P., Huang W. X., Surf. Sci., 2012, 606, 1227—1232 |
| [37] | Guo F. Y., Long G. G., Zhang J., Zhang Z., Liu C. H., Yu K., Appl. Surf. Sci., 2015, 324, 584—589 |
| [1] | 何鸿锐, 夏文生, 张庆红, 万惠霖. 羟基氧化铟团簇与二氧化碳和甲烷作用的密度泛函理论研究[J]. 高等学校化学学报, 2022, 43(8): 20220196. |
| [2] | 姜宏斌, 代文臣, 张娆, 徐晓晨, 陈捷, 杨光, 杨凤林. Co3O4/UiO-66@α-Al2O3陶瓷膜对VOCs废气的分离催化性能[J]. 高等学校化学学报, 2022, 43(6): 20220025. |
| [3] | 戴卫, 侯华, 王宝山. 七氟异丁腈负离子结构与反应活性的理论研究[J]. 高等学校化学学报, 2022, 43(6): 20220044. |
| [4] | 郝宏蕾, 孟繁雨, 李若钰, 李迎秋, 贾明君, 张文祥, 袁晓玲. 生物质基氮掺杂多孔炭材料的制备及对水中亚甲基蓝的吸附性能[J]. 高等学校化学学报, 2022, 43(6): 20220055. |
| [5] | 黄汉浩, 卢湫阳, 孙明子, 黄勃龙. 石墨炔原子催化剂的崭新道路:基于自验证机器学习方法的筛选策略[J]. 高等学校化学学报, 2022, 43(5): 20220042. |
| [6] | 王红宁, 黄丽, 清江, 马腾洲, 蒋伟, 黄维秋, 陈若愚. 香蒲基生物炭的活化及对VOCs吸附的应用[J]. 高等学校化学学报, 2022, 43(4): 20210824. |
| [7] | 陈潇禄, 袁珍闫, 仲迎春, 任浩. 机械球磨制备三苯胺基PAF-106s及C2烃吸附性质[J]. 高等学校化学学报, 2022, 43(3): 20210771. |
| [8] | 孟祥龙, 杨歌, 郭海玲, 刘晨光, 柴永明, 王纯正, 郭永梅. 纳米分子筛的合成及硫化氢吸附性能[J]. 高等学校化学学报, 2022, 43(3): 20210687. |
| [9] | 靳科研, 白璞, 李小龙, 张佳楠, 闫文付. 新型Mg-Al吸附剂去除压水堆核电厂废水中高浓度硼[J]. 高等学校化学学报, 2022, 43(2): 20210516. |
| [10] | 谭乐见, 仲宣树, 王锦, 刘宗建, 张爱英, 叶霖, 冯增国. β-环糊精的低临界溶解温度现象及其在有序纳米孔道片晶制备中的应用[J]. 高等学校化学学报, 2022, 43(11): 20220405. |
| [11] | 刘洋, 李旺昌, 张竹霞, 王芳, 杨文静, 郭臻, 崔鹏. Sc3C2@C80与[12]CPP纳米环之间非共价相互作用的理论研究[J]. 高等学校化学学报, 2022, 43(11): 20220457. |
| [12] | 郑美琪, 毛方琪, 孔祥贵, 段雪. 类水滑石材料在核废水处理领域的应用[J]. 高等学校化学学报, 2022, 43(10): 20220456. |
| [13] | 周成思, 赵远进, 韩美晨, 杨霞, 刘晨光, 贺爱华. 硅烷类外给电子体对丙烯-丁烯序贯聚合的调控作用[J]. 高等学校化学学报, 2022, 43(10): 20220290. |
| [14] | 王园月, 安梭梭, 郑旭明, 赵彦英. 5-巯基-1, 3, 4-噻二唑-2-硫酮微溶剂团簇的光谱和理论计算研究[J]. 高等学校化学学报, 2022, 43(10): 20220354. |
| [15] | 田晓康, 张青松, 杨舒淋, 白洁, 陈冰洁, 潘杰, 陈莉, 危岩. 微生物发酵诱导多孔材料: 制备方法和应用[J]. 高等学校化学学报, 2022, 43(10): 20220216. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||