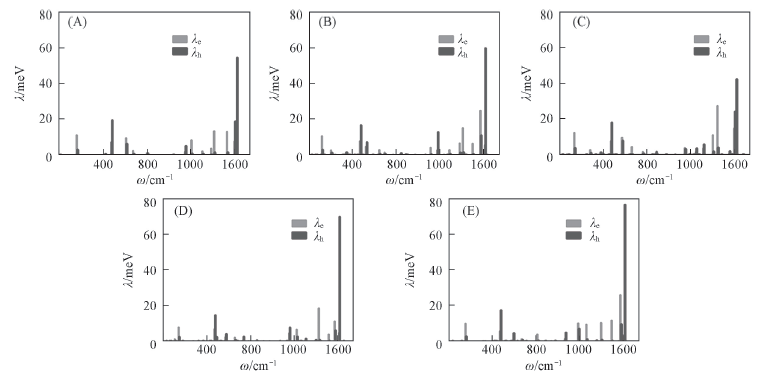
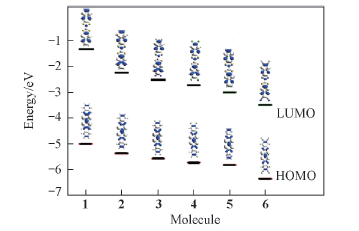
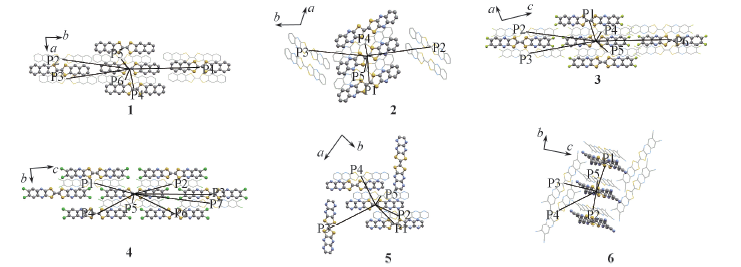
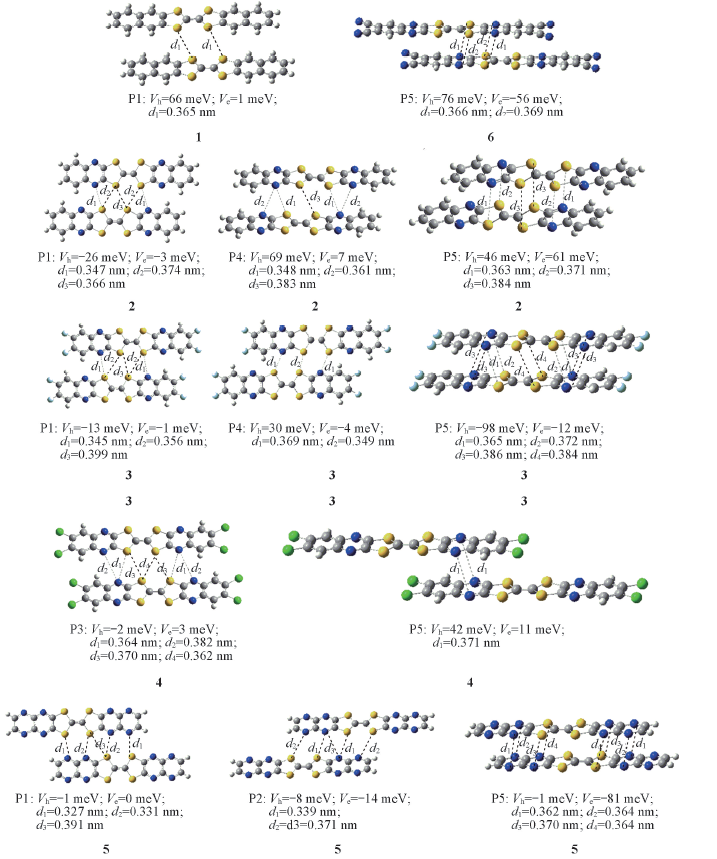
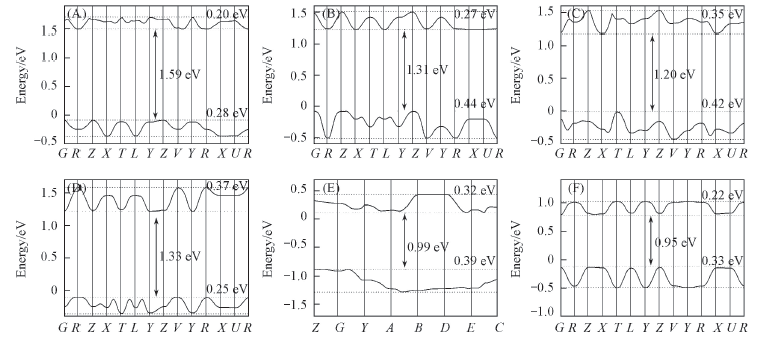
| [1] |
Horowitz G., Fichou D., Peng X., Xu Z., Garnier F., Solid State. Communications, 1989, 72(4), 381—384
|
| [2] |
Facchetti A., Mushrush M., Yoon M. H., Hutchison G. R., Ratner M. A., Marks T. J., J. Am. Chem. Soc., 2004, 126(42), 13859—13874
|
| [3] |
Torrent M., Hadley M. P., Crivillers N., Veciana J., Rovira C., Chem. Phys. Chem., 2006, 7(1), 86—88
|
| [4] |
Jiang H., Yang X., Wang E., Fu Y., Liu Y., Li H., Cui Z., Liu Y., Hu W., Synthetic Metals, 2011, 161(1/2), 136—142
|
| [5] |
Ruiz R., Papadimitratos A., Mayer A. C., Malliaras G. G., Advanced Materials, 2005, 17(14), 1795—1798
|
| [6] |
Yoon M. H., Facchetti A., Stern C. E., Marks T. J., J. Am. Chem. Soc., 2006, 128(17), 5792—5801
|
| [7] |
Marks T. J., Accounts of Chemical Research, 2011, 44(7), 501—510
|
| [8] |
Singh T. B., Erten S., Günes S., Zafer C., Turkmen G., Kuban B., Teoman Y., Sariciftci N., Icli S., Org. Electron., 2006, 7(6), 480—489
|
| [9] |
Leufgen M., Rost O., Gould C., Schmidt G., Geurts J., Molenkamp L. W., Oxtoby N. S., Mas-Torrent M., Crivillers N., Veciana J., Rovira C., Org. Electron., 2008, 9, 1101—1106
|
| [10] |
Ando S., Murakami R., Nishida J. I., Tada H., Inoue Y., Tokito S., Yamashita Y., J. Am. Chem. Soc., 2005, 127(43), 14996—14997
|
| [11] |
Guøgano X., Kanibolotsky A. L., Blum C., Mertens S. F. L., Liu S. X., Neels A., Hagemann H., Skabara P. J., Leutwyler S., Wandlowski T., Hauser A., Decurtins S., Chemistry: A European Journal, 2009, 15(1), 63—66
|
| [12] |
Li H. X., Zheng R. H., Shi Q., Phys. Chem. Chem. Phys., 2011, 13, 5642—5650
|
| [13] |
Geng Y., Wu S. X., Li H. B., Tang X. D., Wu Y., Su Z. M., Liao Y., Journal of Materials Chemistry, 2011, 21(39), 15558—15566
|
| [14] |
Jortner J., Ben-Reuven A., Chemical Physics Letters, 1976, 41(3), 401—406
|
| [15] |
Brunschwig B. S., Logan J., Newton M. D., Sutin N., J. Am. Chem. Soc., 1980, 102(18), 5798—5809
|
| [16] |
Siders P., Marcus R., J. Am. Chem. Soc., 1981, 103(4), 748—752
|
| [17] |
Newton M. D., Sutin N., Annual Review of Physical Chemistry, 1984, 35(1), 437—480
|
| [18] |
Norton J. E., Brédas J. L., J. Am. Chem. Soc., 2008, 130(37), 12377—12384
|
| [19] |
Tang X. D., Geng Y., Liao Y., Su Z. M., Chem. J. Chinese Universities, 2010, 31(4), 766—771
|
|
(汤肖丹, 耿允, 廖奕, 苏忠民. 高等学校化学学报, 2010, 31(4), 766—771)
|
| [20] |
Te Velde G., Bickelhaupt F. M., Baerends E. J., Fonseca Guerra C., van Gisbergen S. J., Snijders J. G., Ziegler T., Journal of Computational Chemistry, 2001, 22(9), 931—967
|
| [21] |
Valeev E. F., Coropceanu V., da Silva Filho D. A., Salman S., Brédas J. L., J. Am. Chem. Soc., 2006, 128(30), 9882—9886
|
| [22] |
Schein L., McGhie A., Physical Review B, 1979, 20, 1631—1639
|
| [23] |
Becke A. D., Physical Review A, 1988, 38(6), 3098
|
| [24] |
Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam N. J., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J., Gaussian 09, Revision D.01, Gaussian Inc.,Wallingford CT, 2009
|
| [25] |
Scott A. P., Radom L., Journal of Physical Chemistry, 1996, 100(41), 16502—16513
|
| [26] |
Kresse G., Furthmüller J., Computational Materials Science, 1996, 6(1), 15—50
|
| [27] |
Kresse G., Hafner J., Physical Review B, 1994, 49(20), 14251
|
| [28] |
Perdew J., Phys. Rev. Lett., 1996, 77, 3865—3868
|
| [29] |
Tang X. D., Liao Y., Geng H., Shuai Z. G., Journal of Materials Chemistry, 2012, 22(35), 18181—18191
|
| [30] |
Lv A., Puniredd S. R., Zhang J., Li Z., Zhu H., Jiang W., Dong H., He Y., Jiang L., Li Y., Pisula W., Meng Q., Hu W., Wang Z., Advanced Materials, 2012, 24(19), 2626—2630
|
| [31] |
Wang C. L., Dong H. L., Hu W. P., Chemical Reviews, 2011, 112(4), 2208—2267
|
| [32] |
Naraso Nishida J. I., Kumaki D., Tokito S., Yamashita Y., J. Am. Chem. Soc., 2006, 128(43), 9598—9599
|
| [33] |
Koh S. E., Delley B., Medvedeva J. E., Facchetti A., Freeman A. J., Marks T. J., Ratner M. A., Journal of Physical Chemistry B, 2006, 110(48), 24361—24370
|
|
(Ed.: Y, Z)
|
 )
)