

高等学校化学学报 ›› 2017, Vol. 38 ›› Issue (9): 1568.doi: 10.7503/cjcu20170213
段永斌1, 殷燕1( ), 孟凡丽1, 赵连花1, 刘玉坤1, 袁哲1, 冯阳波2
), 孟凡丽1, 赵连花1, 刘玉坤1, 袁哲1, 冯阳波2
收稿日期:2017-04-11
出版日期:2017-09-10
发布日期:2017-08-22
作者简介:联系人简介: 殷 燕, 女, 博士, 副教授, 主要从事药物化学方面的研究. E-mail:基金资助:DUAN Yongbin1, YIN Yan1,*, MENG Fanli1, ZHAO Lianhua1, LIU Yukun1, YUAN Zhe1, FENG Yangbo2
Received:2017-04-11
Online:2017-09-10
Published:2017-08-22
Contact:
YIN Yan
E-mail:yinyan@sit.edu.cn
Supported by:摘要:
以3个已报道的苯并噻唑类Rho关联含卷曲螺旋蛋白激酶(ROCK)抑制剂(化合物1~3)为研究对象, 经分子动力学模拟获得其在ROCK2蛋白结合口袋中的稳定结合构象, 通过分子对接结果从氨基酸角度初步揭示了此类抑制剂的结构-活性关系(SAR); 然后, 对这3个抑制剂进行MM/GBSA结合自由能(ΔGbind)研究, 结合自由能计算可知ΔGbind与化合物活性之间具有良好的相关性, 且范德华作用能(ΔGVDW)对ΔGbind的贡献最大. 通过自由能分解获得了对于高活性抑制剂具有重要影响的关键残基. 最后, 根据分子对接和自由能研究结果设计并合成了3类新型苯并噻唑类似物(D1~D10). 生物学评价结果表明, 这10个化合物分别具有11~288 nmol/L(ROCK1)和2~105 nmol/L(ROCK2)的抑制活性. 其中, 化合物D3~D5在人肝微粒体代谢研究中展现出比已报道化合物更高的代谢稳定性. 本研究不仅为高活性ROCK抑制剂的设计提供了理论指导, 也为ROCK的应用研究提供了一系列结构新颖的高活性抑制剂.
中图分类号:
TrendMD:
段永斌, 殷燕, 孟凡丽, 赵连花, 刘玉坤, 袁哲, 冯阳波. 基于分子对接和自由能计算的高活性苯并噻唑类ROCK抑制剂的设计、 合成和生物学评价. 高等学校化学学报, 2017, 38(9): 1568.
DUAN Yongbin, YIN Yan, MENG Fanli, ZHAO Lianhua, LIU Yukun, YUAN Zhe, FENG Yangbo. Design, Synthesis and Biological Evaluation of Benzothiazoles as Highly Potent ROCK Inhibitors Through Molecular Docking and Free Energy Calculations†. Chem. J. Chinese Universities, 2017, 38(9): 1568.
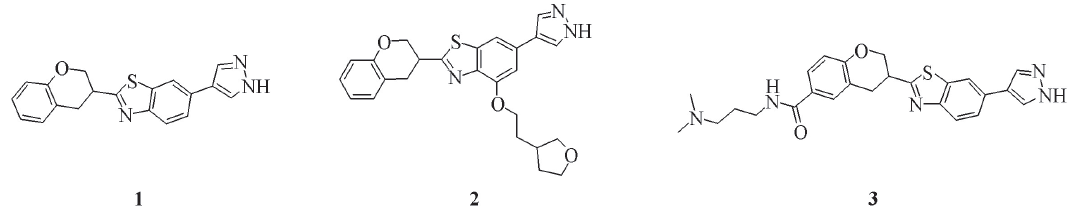
Fig.1 Structures of benzothiazole-based ROCK inhibitors 1—3
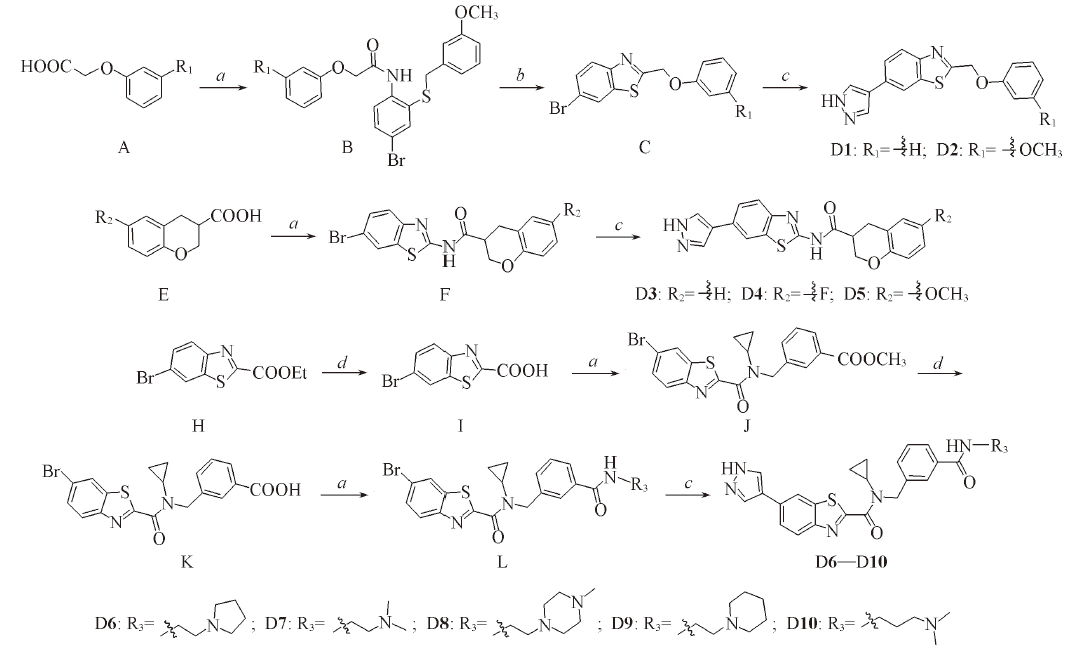
Scheme 1 Synthetic routes of compounds D1—D10Reagents and conditions: a. HATU, amine, DIEA, DMF, r. t. ; b. HOAc, TFA, 100 ℃; c. Pd(PPh3)4, K2CO3, 2-aminopyrid-in-4-ylboronic acid, dioxane/H2O, 95 ℃; d. saturated NaOH solution.
| Compd. | Appearance | Yield(%) | m.p./℃ | HRMS(calcd.)[M+H]+ | Elemental analysis(%, calcd.) | ||
|---|---|---|---|---|---|---|---|
| C | H | O | |||||
| D1 | White solid | 52% | 204—205 | 308.0872(308.0858) | 66.51(66.43) | 4.36(4.26) | 13.45(13.62) |
| D2 | White solid | 50% | 214—215 | 338.0947(338.0963) | 64.12(64.08) | 4.36(4.48) | 12.71(12.49) |
| D3 | White solid | 70% | 234—235 | 337.1061(337.1072) | 63.99(63.81) | 4.36(4.28) | 14.69(14.81) |
| D4 | White solid | 68% | 248—249 | 395.0959(395.0978) | 60.99(60.90) | 3.96(3.83) | 14.61(14.42) |
| D5 | White solid | 67% | 281—282 | 407.1193(407.1178) | 62.19(62.05) | 4.69(4.46) | 13.92(13.74) |
| D6 | White solid | 31% | 238—239 | 515.2241(515.2229) | 65.51(65.35) | 5.66(5.88) | 16.15(16.13) |
| D7 | White solid | 32% | 244—245 | 489.2059(489.2073) | 63.98(63.91) | 5.53(5.78) | 17.06(17.12) |
| D8 | White solid | 30% | 223—224 | 544.2503(544.2495) | 64.98(64.03) | 6.83(6.62) | 18.06(18.00) |
| D9 | White solid | 31% | 216—217 | 529.2359(529.2386) | 65.98(65.88) | 6.02(6.10) | 15.83(15.90) |
| D10 | White solid | 34% | 254—255 | 503.2247(503.2229) | 64.43(64.52) | 5.97(6.02) | 16.83(16.70) |
Table 1 Appearance, yields, melting points, HRMS and elemental analysis data of compounds D1—D10
| Compd. | Appearance | Yield(%) | m.p./℃ | HRMS(calcd.)[M+H]+ | Elemental analysis(%, calcd.) | ||
|---|---|---|---|---|---|---|---|
| C | H | O | |||||
| D1 | White solid | 52% | 204—205 | 308.0872(308.0858) | 66.51(66.43) | 4.36(4.26) | 13.45(13.62) |
| D2 | White solid | 50% | 214—215 | 338.0947(338.0963) | 64.12(64.08) | 4.36(4.48) | 12.71(12.49) |
| D3 | White solid | 70% | 234—235 | 337.1061(337.1072) | 63.99(63.81) | 4.36(4.28) | 14.69(14.81) |
| D4 | White solid | 68% | 248—249 | 395.0959(395.0978) | 60.99(60.90) | 3.96(3.83) | 14.61(14.42) |
| D5 | White solid | 67% | 281—282 | 407.1193(407.1178) | 62.19(62.05) | 4.69(4.46) | 13.92(13.74) |
| D6 | White solid | 31% | 238—239 | 515.2241(515.2229) | 65.51(65.35) | 5.66(5.88) | 16.15(16.13) |
| D7 | White solid | 32% | 244—245 | 489.2059(489.2073) | 63.98(63.91) | 5.53(5.78) | 17.06(17.12) |
| D8 | White solid | 30% | 223—224 | 544.2503(544.2495) | 64.98(64.03) | 6.83(6.62) | 18.06(18.00) |
| D9 | White solid | 31% | 216—217 | 529.2359(529.2386) | 65.98(65.88) | 6.02(6.10) | 15.83(15.90) |
| D10 | White solid | 34% | 254—255 | 503.2247(503.2229) | 64.43(64.52) | 5.97(6.02) | 16.83(16.70) |
| Compd. | 1H NMR(400 MHz, DMSO-d6), δ |
|---|---|
| D1 | 9.68(s, 1H, NH), 8.46—8.45(m, 1H), 8.20—8.19(m, 1H), 8.09—8.07(m, 1H), 7.90—7.89(m, 1H), 7.65—7.52(m, 4H), 7.32—7.23(m, 2H), 4.02(s, 2H, CH3) |
| D2 | 8.43(s, 1H, NH), 8.09—8.02(m, 2H), 7.89—7.84(m, 1H), 7.32—7.24(m, 2H), 6.93—6.86(m, 4H), 4.00(s, 2H, CH2), 3.75(s, 3H, CH3) |
| D3 | 12.94(s, 1H, NH), 12.63(s, 1H, NH), 8.22(s, 1H), 8.14—8.11(m, 1H), 7.74—7.72(m, 2H), 7.64—7.57(m, 1H), 7.16—7.08(m, 2H), 6.89—6.86(m, 1H), 6.80—6.78(m, 1H), 4.48—4.46(m, 1H, CH2), 4.14—4.10(m, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.11—3.01(m, 2H, CH2) |
| D3 | 12.94(s, 1H, NH), 12.63(s, 1H, NH), 8.22(s, 1H), 8.14—8.11(m, 1H), 7.74—7.72(m, 2H), 7.64—7.57(m, 1H), 7.16—7.08(m, 2H), 6.89—6.86(m, 1H), 6.80—6.78(m, 1H), 4.48—4.46(m, 1H, CH2), 4.14—4.10(m, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.11—3.01(m, 2H, CH2) |
| D4 | 12.63(s, 1H, NH), 8.22(s, 1H, NH), 8.21—8.05(m, 2H), 7.74—7.68(m, 2H), 7.05—7.02(m, 1H), 6.96—6.91(m, 2H), 6.82—6.79(m, 1H), 4.44(dd, J=10.8, 2.4 Hz, 1H, CH2), 4.13(dd, J=10.8, 8.4 Hz, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.07—3.02(m, 2H, CH2) |
| D5 | 12.61(s, 1H, NH), 8.25(s, 1H, NH), 8.22—8.21(m, 1H), 8.13—8.10(m, 2H), 7.74—7.68(m, 2H), 6.74—6.67(m, 3H), 4.40(dd, J=10.8, 2.0 Hz, 1H, CH2), 4.06(dd, J=10.8, 8.8 Hz, 1H, CH2), 3.69(s, 3H, OCH3), 3.09—2.96(m, 3H, CH, CH2) |
| D6 | 9.35(s, 1H, NH), 8.74—8.73(m, 1H, NH), 8.44—8.43(m, 1H), 8.19—8.07(m, 3H), 7.88—7.80(m, 2H), 7.55—7.54(m, 2H), 4.80(s, 2H, CH2), 3.58—3.56(m, 5H, CH, CH2), 3.34—3.30(m, 2H, CH2), 3.10—3.06(m, 2H, CH2), 1.85—1.84(m, 4H, CH2), 0.79—0.69(m, 4H, CH2) |
| D7 | 9.19(s, 1H, NH), 8.65—8.64(m, 1H), 8.37—8.36(m, 1H), 8.13—8.12(m, 1H), 8.02—8.00(m, 1H), 7.81—7.73(m, 3H), 7.48—7.47(m, 2H), 4.76(s, 2H), 3.62—3.61(m, 1H), 3.53—3.52(m, 2H), 3.20—3.10(m, 2H), 2.78(s, 3H), 2.77(s, 3H), 1.82—1.79(m, 2H), 0.83—0.79(m, 2H) |
| D8 | 9.17(s, 1H, NH), 8.6—8.58(m, 1H), 8.33—8.31(m, 1H), 8.10—8.08(m, 1H), 8.05—8.03(m, 1H), 7.85—7.76(m, 3H), 7.45—7.43(m, 2H), 4.72(s, 2H, CH2), 3.62—3.61(m, 1H, CH), 3.53—3.52(m, 6H, CH2), 3.20—3.10(m, 6H, CH2), 2.78(s, 3H, CH3), 1.80—1.79(m, 2H, CH2), 0.87—0.84(m, 2H, CH2) |
| D9 | 9.32(s, 1H, NH), 8.71—8.69(m, 1H), 8.47—8.45(m, 1H), 8.10—8.07(m, 3H), 7.88—7.64(m, 4H), 4.81(s, 2H, CH2), 3.65—3.63(m, 1H, CH), 3.58—3.56(m, 2H, CH2), 3.34—3.30(m, 2H, CH2), 3.10—3.06(m, 2H, CH2), 2.01—1.99(m, 2H, CH2), 1.85—1.84(m, 4H, CH2), 0.79—0.69(m, 6H, CH2) |
| D10 | 9.10(s, 1H, NH), 8.78—8.79(m, 1H), 8.34—8.30(m, 1H), 8.10—7.98(m, 2H), 7.85—7.74(m, 3H), 7.55—7.47(m, 2H), 4.75(s, 2H, CH2), 3.67—3.65(m, 1H, CH), 3.60—3.58(m, 2H, CH2), 3.56—3.54(m, 2H, CH2), 3.19—3.08(m, 2H, CH2), 2.80(s, 3H, CH3), 2.78(s, 3H, CH3), 1.80—1.78(m, 2H, CH2), 1.45—1.41(m, 2H, CH2), 0.84—0.83(m, 2H, CH2) |
Table 2 1H NMR data of compounds D1—D10
| Compd. | 1H NMR(400 MHz, DMSO-d6), δ |
|---|---|
| D1 | 9.68(s, 1H, NH), 8.46—8.45(m, 1H), 8.20—8.19(m, 1H), 8.09—8.07(m, 1H), 7.90—7.89(m, 1H), 7.65—7.52(m, 4H), 7.32—7.23(m, 2H), 4.02(s, 2H, CH3) |
| D2 | 8.43(s, 1H, NH), 8.09—8.02(m, 2H), 7.89—7.84(m, 1H), 7.32—7.24(m, 2H), 6.93—6.86(m, 4H), 4.00(s, 2H, CH2), 3.75(s, 3H, CH3) |
| D3 | 12.94(s, 1H, NH), 12.63(s, 1H, NH), 8.22(s, 1H), 8.14—8.11(m, 1H), 7.74—7.72(m, 2H), 7.64—7.57(m, 1H), 7.16—7.08(m, 2H), 6.89—6.86(m, 1H), 6.80—6.78(m, 1H), 4.48—4.46(m, 1H, CH2), 4.14—4.10(m, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.11—3.01(m, 2H, CH2) |
| D3 | 12.94(s, 1H, NH), 12.63(s, 1H, NH), 8.22(s, 1H), 8.14—8.11(m, 1H), 7.74—7.72(m, 2H), 7.64—7.57(m, 1H), 7.16—7.08(m, 2H), 6.89—6.86(m, 1H), 6.80—6.78(m, 1H), 4.48—4.46(m, 1H, CH2), 4.14—4.10(m, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.11—3.01(m, 2H, CH2) |
| D4 | 12.63(s, 1H, NH), 8.22(s, 1H, NH), 8.21—8.05(m, 2H), 7.74—7.68(m, 2H), 7.05—7.02(m, 1H), 6.96—6.91(m, 2H), 6.82—6.79(m, 1H), 4.44(dd, J=10.8, 2.4 Hz, 1H, CH2), 4.13(dd, J=10.8, 8.4 Hz, 1H, CH2), 3.25—3.23(m, 1H, CH), 3.07—3.02(m, 2H, CH2) |
| D5 | 12.61(s, 1H, NH), 8.25(s, 1H, NH), 8.22—8.21(m, 1H), 8.13—8.10(m, 2H), 7.74—7.68(m, 2H), 6.74—6.67(m, 3H), 4.40(dd, J=10.8, 2.0 Hz, 1H, CH2), 4.06(dd, J=10.8, 8.8 Hz, 1H, CH2), 3.69(s, 3H, OCH3), 3.09—2.96(m, 3H, CH, CH2) |
| D6 | 9.35(s, 1H, NH), 8.74—8.73(m, 1H, NH), 8.44—8.43(m, 1H), 8.19—8.07(m, 3H), 7.88—7.80(m, 2H), 7.55—7.54(m, 2H), 4.80(s, 2H, CH2), 3.58—3.56(m, 5H, CH, CH2), 3.34—3.30(m, 2H, CH2), 3.10—3.06(m, 2H, CH2), 1.85—1.84(m, 4H, CH2), 0.79—0.69(m, 4H, CH2) |
| D7 | 9.19(s, 1H, NH), 8.65—8.64(m, 1H), 8.37—8.36(m, 1H), 8.13—8.12(m, 1H), 8.02—8.00(m, 1H), 7.81—7.73(m, 3H), 7.48—7.47(m, 2H), 4.76(s, 2H), 3.62—3.61(m, 1H), 3.53—3.52(m, 2H), 3.20—3.10(m, 2H), 2.78(s, 3H), 2.77(s, 3H), 1.82—1.79(m, 2H), 0.83—0.79(m, 2H) |
| D8 | 9.17(s, 1H, NH), 8.6—8.58(m, 1H), 8.33—8.31(m, 1H), 8.10—8.08(m, 1H), 8.05—8.03(m, 1H), 7.85—7.76(m, 3H), 7.45—7.43(m, 2H), 4.72(s, 2H, CH2), 3.62—3.61(m, 1H, CH), 3.53—3.52(m, 6H, CH2), 3.20—3.10(m, 6H, CH2), 2.78(s, 3H, CH3), 1.80—1.79(m, 2H, CH2), 0.87—0.84(m, 2H, CH2) |
| D9 | 9.32(s, 1H, NH), 8.71—8.69(m, 1H), 8.47—8.45(m, 1H), 8.10—8.07(m, 3H), 7.88—7.64(m, 4H), 4.81(s, 2H, CH2), 3.65—3.63(m, 1H, CH), 3.58—3.56(m, 2H, CH2), 3.34—3.30(m, 2H, CH2), 3.10—3.06(m, 2H, CH2), 2.01—1.99(m, 2H, CH2), 1.85—1.84(m, 4H, CH2), 0.79—0.69(m, 6H, CH2) |
| D10 | 9.10(s, 1H, NH), 8.78—8.79(m, 1H), 8.34—8.30(m, 1H), 8.10—7.98(m, 2H), 7.85—7.74(m, 3H), 7.55—7.47(m, 2H), 4.75(s, 2H, CH2), 3.67—3.65(m, 1H, CH), 3.60—3.58(m, 2H, CH2), 3.56—3.54(m, 2H, CH2), 3.19—3.08(m, 2H, CH2), 2.80(s, 3H, CH3), 2.78(s, 3H, CH3), 1.80—1.78(m, 2H, CH2), 1.45—1.41(m, 2H, CH2), 0.84—0.83(m, 2H, CH2) |
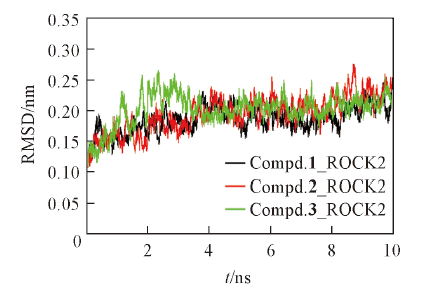
Fig.2 RMSD of the backbone Cα atoms vs. simulation time
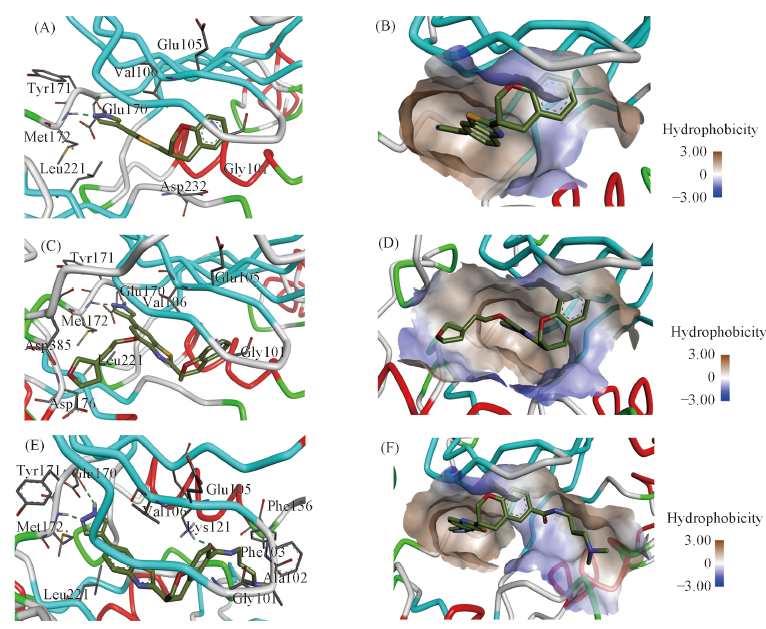
Fig.3 Docking results of compounds 1—3 and the catalytic domain of ROCK2(A), (C) and (E): interaction of compounds 1—3 and ROCK2, respectively; (B), (D) and (F): the hydrophobic pocket of ROCK2 with compounds 1—3, respectively.
| Compd. | ΔEVDW/ (kJ·mol-1) | ΔGSA/ (kJ·mol-1) | ΔEELE/ (kJ·mol-1) | ΔGGB/ (kJ·mol-1) | ΔGbind/ (kJ·mol-1) | IC50/ (nmol·L-1) |
|---|---|---|---|---|---|---|
| 1 | -181.4458 | -21.8627 | -152.2825 | 170.4642 | -185.1268 | 11 |
| 2 | -214.9761 | -26.5596 | -67.0682 | 140.4793 | -168.1246 | 500 |
| 3 | -226.3453 | -26.0829 | -141.0213 | 191.2943 | -202.1552 | 0.9 |
Table 3 Predicted binding free energies and individual energy components
| Compd. | ΔEVDW/ (kJ·mol-1) | ΔGSA/ (kJ·mol-1) | ΔEELE/ (kJ·mol-1) | ΔGGB/ (kJ·mol-1) | ΔGbind/ (kJ·mol-1) | IC50/ (nmol·L-1) |
|---|---|---|---|---|---|---|
| 1 | -181.4458 | -21.8627 | -152.2825 | 170.4642 | -185.1268 | 11 |
| 2 | -214.9761 | -26.5596 | -67.0682 | 140.4793 | -168.1246 | 500 |
| 3 | -226.3453 | -26.0829 | -141.0213 | 191.2943 | -202.1552 | 0.9 |
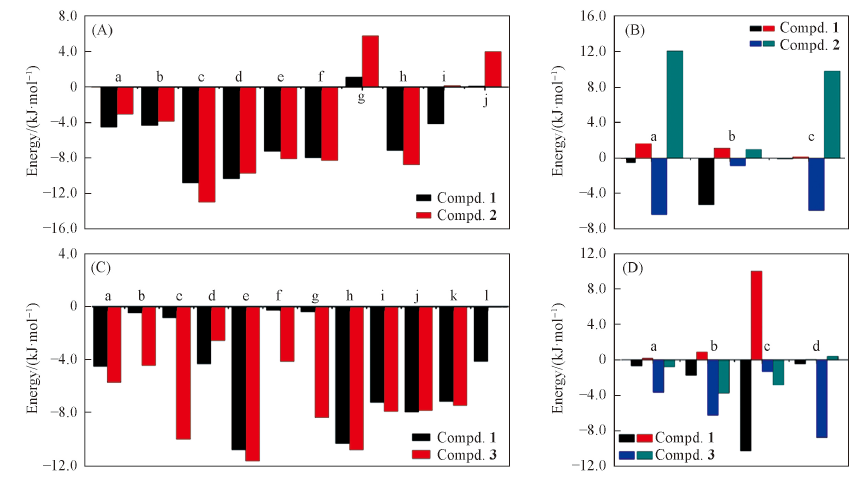
Fig.4 Contribution of key residues to binding energy(A, C) and individual energy terms(B, D)
| Compd. | Structure | IC50/(nmol·L-1) | ||
|---|---|---|---|---|
| ROCK1 | ROCK2 | |||
| 1 | 64 | 11 | 18 | |
| 3 | 2 | 0.9 | 34 | |
| D1 | 72 | 44 | 60 | |
| D2 | 64 | 42 | 19 | |
| D3 | 72 | 14 | 45.9 | |
| D4 | 171 | 22 | 56.4 | |
| D5 | 288 | 105 | 91 | |
| D6 | 60 | 21 | 30 | |
| D7 | 95 | 35 | 12 | |
| D8 | 11 | 2 | 9 | |
| D9 | 58 | 37 | 13 | |
| D10 | 45 | 19 | 45 | |
Table 4 Inhibitory activities and metabolic stabilities of newly designed inhibitors
| Compd. | Structure | IC50/(nmol·L-1) | ||
|---|---|---|---|---|
| ROCK1 | ROCK2 | |||
| 1 | 64 | 11 | 18 | |
| 3 | 2 | 0.9 | 34 | |
| D1 | 72 | 44 | 60 | |
| D2 | 64 | 42 | 19 | |
| D3 | 72 | 14 | 45.9 | |
| D4 | 171 | 22 | 56.4 | |
| D5 | 288 | 105 | 91 | |
| D6 | 60 | 21 | 30 | |
| D7 | 95 | 35 | 12 | |
| D8 | 11 | 2 | 9 | |
| D9 | 58 | 37 | 13 | |
| D10 | 45 | 19 | 45 | |
| [1] | Nakagawa O., Fujisawa K., Ishizaki T., Saito Y., Nakao K., Narumiya S., FEBS Lett., 1996, 392(2), 189—193 |
| [2] | Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Narumiya S., Nature,1997, 389(6654), 990—994 |
| [3] | Sato M., Tani E., Fujikawa H., Kaibuchi K., Circ. Res., 2000, 87(3), 195—200 |
| [4] | Kajikawa M., Noma K., Nakashima A., Maruhashi T., Iwamoto Y., Matsumoto T., Aibara Y., Hypertension,2015, 66(4), 892—899 |
| [5] | del Re D. P., Miyamoto S., Brown J. H., J. Biol. Chem., 2007, 282(11), 8069—8078 |
| [6] | Fernández-Gamba A., Leal M. C., Maarouf C. L., Richter-Landsberg C., Wu T., Morelli L., Castaño E. M., J. Neurochem., 2012, 121(6), 985—995 |
| [7] | Challa P., Arnold J. J., Expert Opin. Invest. Drugs, 2014, 23(1), 81—95 |
| [8] | Surma M., Wei L., Shi J., Future Cardiol., 2011, 7(5), 657—671 |
| [9] | Huentelman M. J., Stephan D. A., Talboom J., Corneveaux J. J., Reiman D. M., Gerber J. D., Bimonte-Nelson H. A., Behav. Neurosci., 2009, 123(1), 218—223 |
| [10] | Rath N., Olson M. F., EMBO Rep., 2012, 13(10), 900—908 |
| [11] | Shimizu Y., Dobashi K., Sano T., Yamada M., Int. J. Immunopathol. Pharmacol., 2014, 27(1), 37—44 |
| [12] | Schirok H., Paulsen H., Kroh W., Chen G., Gao P., Org. Process Res. Dev., 2009, 13(2), 168—173 |
| [13] | Shen M., Zhou S., Li Y., Pan P., Zhang L., Hou T., Mol. BioSyst., 2013, 9(3), 361—374 |
| [14] | Sumi K., Inoue Y., Nishio M., Naito Y., Hosoya T., Suzuki M., Hidaka H., Bioorg. Med. Chem. Lett., 2014, 24(3), 831—834 |
| [15] | Yin Y., Lin L., Ruiz C., Khan S., Cameron M. D., Grant W., LoGrasso P. V., J. Med. Chem., 2013, 56(9), 3568—3581 |
| [16] | Green J., Cao J., Bandarage U. K., Gao H., Court J., Marhefka C., Shah S., J. Med. Chem., 2015, 58(12), 5028—5037 |
| [17] | Boland S., Bourin A., Alen J., Geraets J., Schroeders P., Castermans K., Vanormelingen J., J. Med. Chem., 2015, 58(10), 4309—4324 |
| [18] | Ding M., Yin Y., Wu F., Cui J., Zhou H., Sun G., Feng Y., Bioorg. Med. Chem., 2015, 23(10), 2505—2517 |
| [19] | Cui J., Ding M., Deng W., Yin Y., Wang Z., Zhou H., Feng Y., Bioorg. Med. Chem., 2015, 23(23), 7464—7477 |
| [20] | Zhao Z., Cui J., Yin Y., Zhang H., Liu Y., Zeng R., Wu F., Chin. J. Chem., 2016, 34(8), 801—808 |
| [21] | Yin Y., Lin L., Ruiz C., Cameron M. D., Pocas J., Grant W., LoGrasso P., Bioorg. Med. Chem. Lett., 2009, 19(23), 6686—6690 |
| [22] | Huang Y. L., Gao X. F., Chem. J. Chinese Universities, 2016, 37(5), 928—931 |
| (黄义玲, 高雪峰.高等学校化学学报,2016, 37(5), 928—931) | |
| [23] | Li H., Zou H., Liu L., Zhao D., Yang Z., Chem. Res. Chinese Universities, 2017, 33(2), 239—247 |
| [24] | Yang H., Ren Y., Gao X., Gao Y., Chem. Res. Chinese Universities, 2016, 32(6), 973—978 |
| [25] | Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., J. Chem. Theory Comput., 2015, 11(8), 3696—3713 |
| [26] | Homeyer N., Gohlke H., Mol. Inf., 2012, 31(2), 114—122 |
| [27] | Huo S., Wang J., Cieplak P., Kollman P. A., Kuntz I. D., J. Med. Chem., 2002, 45(7), 1412—1419 |
| [28] | Onufriev A., Bashford D., Case D. A., Proteins,2004, 55(2), 383—394 |
| [29] | Weiser J., Shenkin P. S., Still W. C., J. Comput. Chem., 1999, 20(2), 217—230 |
| [30] | Hou T., Wang J., Li Y., Wang W., J. Chem. Inf. Model., 2010, 51(1), 69—82 |
| [31] | Hou T., Li N., Li Y., Wang W., J. Proteome Res., 2012, 11(5), 2982—2995 |
| [32] | Gohlke H., Kiel C., Case D. A., J. Mol. Biol., 2003, 330(4), 891—913 |
| [33] | Yin Y., Ruiz C., Khan S., Cameron M. D., Grant W., Pocas J., Eid N., Park H., Schröter T., LoGrasso P. V., Feng Y. B., J. Med. Chem., 2013, 56(21), 3568—3581 |
| [34] | Kamenecka T., Jiang R., Song X., Duckett D., Chen W., Ling Y. Y., Cameron M. D., J. Med. Chem., 2010, 53(1), 419—431 |
| [35] | Sessions E. H., Yin Y., Bannister T. D., Weiser A., Griffin E., Pocas J., Schröter T., Bioorg. Med. Chem. Lett., 2008, 18(24), 6390—6393 |
| [1] | 高志伟, 李军委, 史赛, 付强, 贾钧儒, 安海龙. 基于分子动力学模拟的TRPM8通道门控特性分析[J]. 高等学校化学学报, 2022, 43(6): 20220080. |
| [2] | 崔韶丽, 张维佳, 邵学广, 蔡文生. 自由能计算揭示苏氨酸对抗冻蛋白与冰晶结合能力的影响[J]. 高等学校化学学报, 2022, 43(3): 20210838. |
| [3] | 胡波, 朱昊辰. 双层氧化石墨烯纳米体系中受限水的介电常数[J]. 高等学校化学学报, 2022, 43(2): 20210614. |
| [4] | 李聪聪, 刘明皓, 韩佳睿, 朱镜璇, 韩葳葳, 李婉南. 基于分子动力学模拟的VmoLac非特异性底物催化活性的理论研究[J]. 高等学校化学学报, 2021, 42(8): 2518. |
| [5] | 雷晓彤, 金怡卿, 孟烜宇. 基于分子模拟方法预测PIP2在双孔钾通道TREK-1上结合位点的研究[J]. 高等学校化学学报, 2021, 42(8): 2550. |
| [6] | 曾永辉, 言天英. 质子水合结构的振动态密度分析[J]. 高等学校化学学报, 2021, 42(6): 1855. |
| [7] | 齐人睿, 李明昊, 常浩, 付学奇, 高波, 韩葳葳, 韩璐, 李婉南. 基于拉伸分子动力学模拟的黄嘌呤氧化酶抑制剂解离途径的理论研究[J]. 高等学校化学学报, 2021, 42(3): 758. |
| [8] | 刘爱清, 徐文生, 徐晓雷, 陈继忠, 安立佳. 高分子/棒状纳米粒子复合物的分子动力学模拟[J]. 高等学校化学学报, 2021, 42(3): 875. |
| [9] | 帅蝶, 赵美娟, 陈丙年, 王力. 4种Keggin型磷钼酸盐对蘑菇酪氨酸酶活性和黑色素生成的抑制及抗氧化作用[J]. 高等学校化学学报, 2021, 42(12): 3579. |
| [10] | 妙孟姚, 郭一畅, 邵学广, 蔡文生. 分子梭协助离子跨膜运输的机理研究[J]. 高等学校化学学报, 2021, 42(10): 3116. |
| [11] | 杨举, 苏丽娇, 李灿花, 鲁佳佳, 杨俊丽, 古捷, 杨丽, 杨丽娟. 甘松新酮/磷酸盐柱[6]芳烃主客体络合行为[J]. 高等学校化学学报, 2021, 42(10): 3099. |
| [12] | 张爱芹, 王嫚, 申刚义, 金军. 多溴联苯醚与人血清白蛋白相互作用的表面等离子体共振及分子对接[J]. 高等学校化学学报, 2020, 41(9): 2054. |
| [13] | 王莲萍,李庆杰,刘晓艳,任跃英,杨秀伟. 基于UFLC-MS/分子模拟计算的吴茱萸醇提取物中胆碱酯酶抑制剂筛选[J]. 高等学校化学学报, 2020, 41(1): 111. |
| [14] | 王晓霞, 马力通, 聂智华, 王正德, 崔金龙, 赵文渊, 赛华征. 多光谱法和分子对接模拟法研究黄腐酸与胃蛋白酶的相互作用[J]. 高等学校化学学报, 2019, 40(9): 1840. |
| [15] | 瞿思颖, 徐沁. 凝血因子Xa的S4口袋部分关键残基对利伐沙班结合的不同作用机制[J]. 高等学校化学学报, 2019, 40(9): 1918. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||