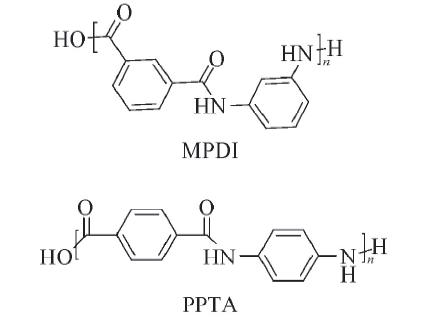
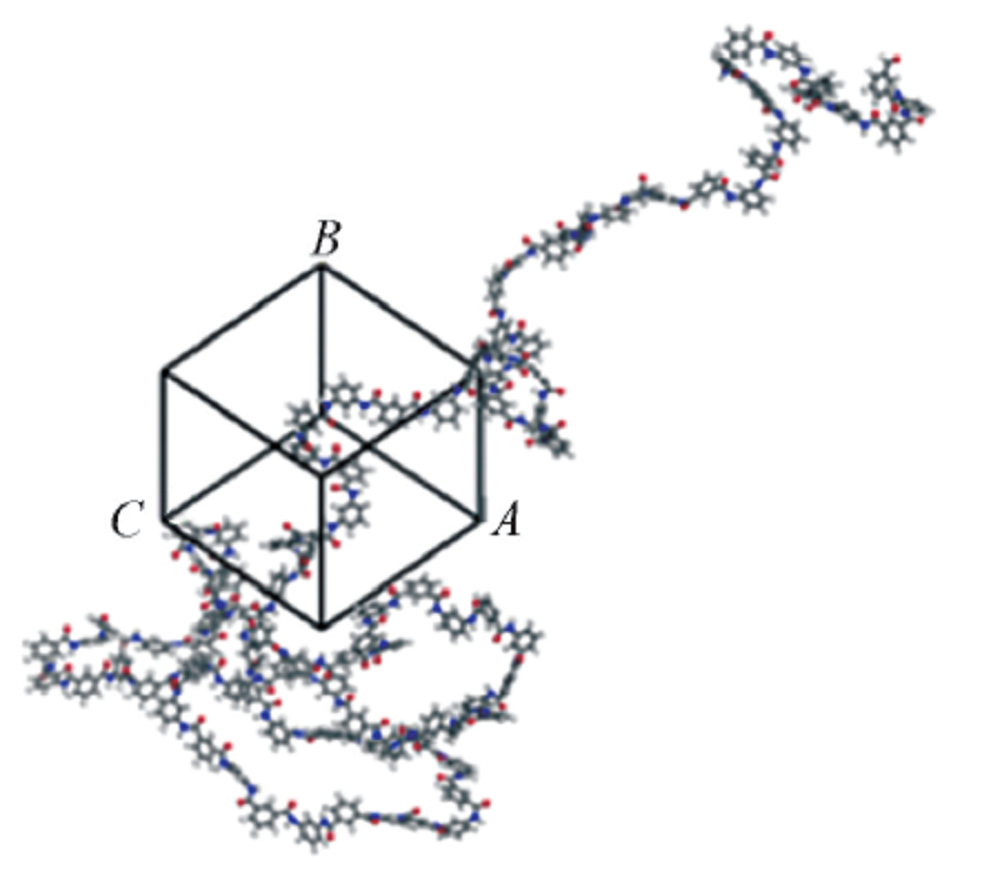
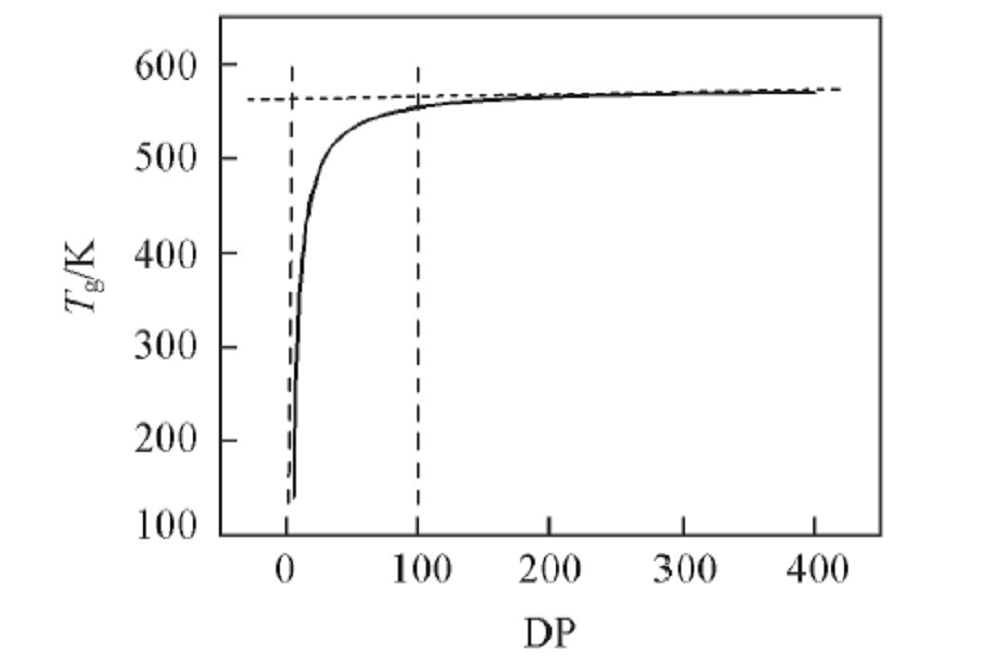
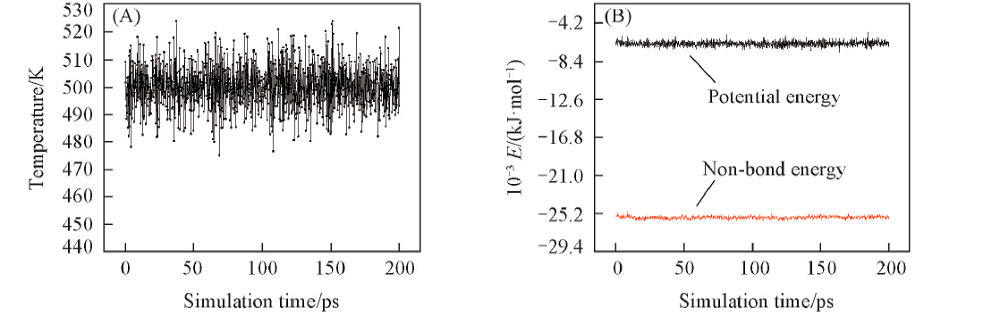
| [1] |
Zhang R., He X. R., Yu H., Polymer Materials Science & Engineering,2015, 31(4), 186—190
|
|
(张睿, 何显儒, 余慧. 高分子材料科学与工程, 2015, 31(4), 186—190)
|
| [2] |
Diego J. A., Canadas J., Mudarra M., Belana J., Polymer,2000, 40(40), 5355—5363
|
| [3] |
Roe R.J., MD Simulation Study of Glass Transition and Short Time Dynamics in Polymer Liquids. In: Atomistic Modeling of Physical Properties, Ed.: Monnerie L., Suter U. W., Springer-Verlag, Berlin Heidelberg, 1994, 116(1), 415—426
|
| [4] |
Han J., Gee R. H., Boyd R. H., Macromolecules,1994, 27(26), 7781—7784
|
| [5] |
Boudouris D., Constantinou L., Panayiotou C., Fluid Phase Equilibria,2000, 167(1), 1—19
|
| [6] |
Wang Y.H., Wang W. H., Zhang Z. Q., Xu L. C., Li P.,European Polymer Journal, 2016, 75, 36—45
|
| [7] |
Cousin T., Galy J., Dupuy J., Polymer,2012, 53(15), 3203—3210
|
| [8] |
García J. M., García F. C., Serna F., Peña J. L. D. L., Progress in Polymer Science,2010, 35(5), 623—686
|
| [9] |
Kakida H., Chatani Y., Tadokoro H., Journal of Polymer Science Polymer Physics Edition,1976, 14(3), 427—435
|
| [10] |
Northolt M. G., van Aartsen J. J., Journal of Polymer Science Part C Polymer Letters,1973, 11(11), 333—337
|
| [11] |
Bicerano J.,Encyclopedia of Polymer Science and Technology, 2006, 655—678
|
| [12] |
Yang Z., Study on The Polycondensation and Spinning Process of Poly(p-phenyleneterephthalamide) Copolymer, Donghua University,Shanghai, 2006
|
|
(杨拯. 聚对苯二甲酰对苯二胺(PPTA)及其共聚物的聚合及纺丝工艺研究, 上海: 东华大学, 2006)
|
| [13] |
Van Krevelen D. W., Hoftyzer P., Journal of Applied Polymer Science,1969, 13(13), 871—881
|
| [14] |
Pant P. V. K., Han J., Smith G. D., Boyd R. H., Journal of Chemical Physics,1993, 99(1), 597—604
|
| [15] |
Abu-Sharkh B. F., Computational & Theoretical Polymer Science, 2001, 11(1), 29—34
|
| [16] |
Sarangapani R., Reddy S. T., Sikder A. K., Journal of Molecular Graphics & Modelling,2015, 57, 114
|
| [17] |
Rigby D., Sun H., Eichinger B. E., Polymer International,2015, 44(3), 311—330
|
| [18] |
Sun H., Journal of Physical Chemistry B, 1998, 102(38), 7338—7364
|
| [19] |
Sacristan J., Mijangos C., Macromolecules,2010, 43(17), 7357—7367
|
| [20] |
Gee R. H., And L. E. F., Cook R. C., Macromolecules,2001, 34(9), 3050—3059
|
| [21] |
Ahmadi A., Freire J. J., Polymer,2009, 50(20), 4973—4978
|
| [22] |
Chang K. S., Huang Y. H., Lee K. R., Tung K. L., Journal of Membrane Science,2010, 354(1), 93—100
|
| [23] |
Fermeglia M., Ferrone M., Pricl S., Fluid Phase Equilibria,2003, 212(1/2), 315—329
|
| [24] |
Ding Y., Wu H. M., Wang M., Liang Z. H., Li Y. C., Journal of Shandong Chemical Industry,2016, 45(11), 74—77
|
|
(丁勇, 吴红枚, 王孟, 梁振辉, 李永成. 山东化工, 2016, 45(11), 74—77)
|
| [25] |
Fu Y. Z., Liu Y. Q., Zhang L. Y., Lan Y. H., Journal of Molecular Science,2009, 25(1), 1—4
|
|
(付一政, 刘亚青, 张丽燕, 兰艳花. 分子科学学报, 2009, 25(1), 1—4)
|
| [26] |
Fu Y. Z., Liu Y. Q., Lan Y. H., Polymer Materials Science & Engineering,2009, 25(10), 53—56
|
|
(付一政, 刘亚青, 兰艳花. 高分子材料科学与工程, 2009, 25(10), 53—56)
|
| [27] |
White R. P., Lipson J. E. G., Macromolecules,2016, 49(11), 3987—4007
|
| [28] |
White R. P., Lipson J. E. G., ACS Macro. Letters,2015, 4(5), 588—592
|
 ), 李惠婷, 李永成, 王宏青, 王孟
), 李惠婷, 李永成, 王宏青, 王孟