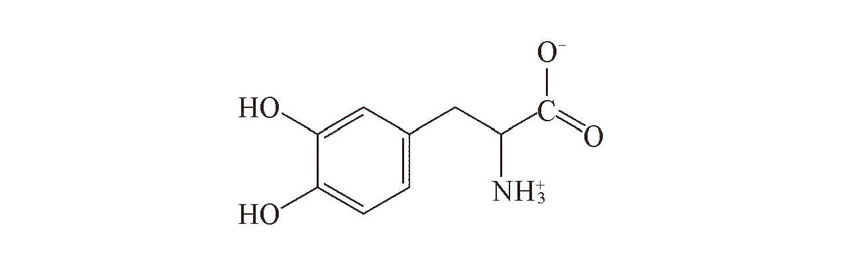
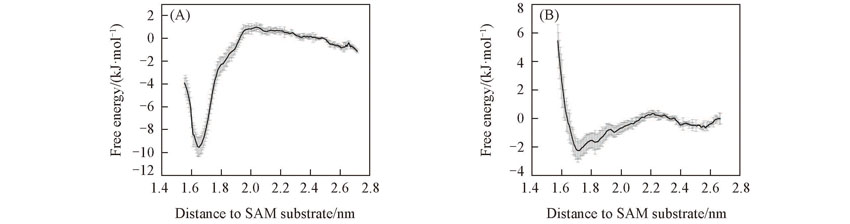
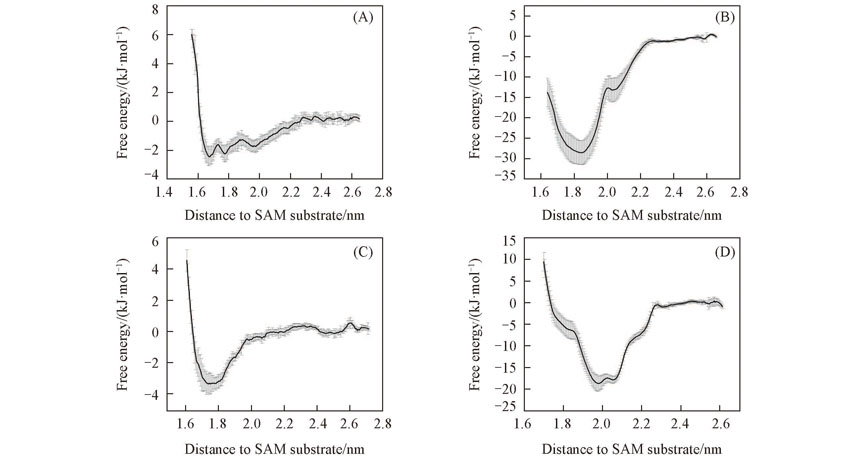
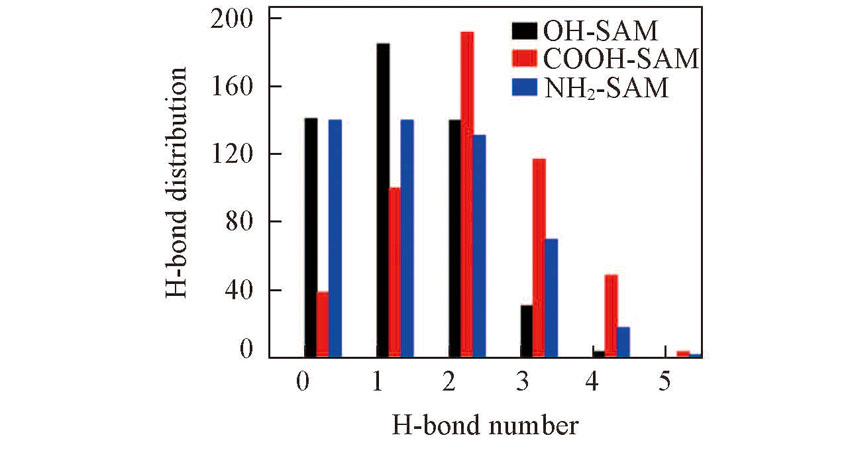
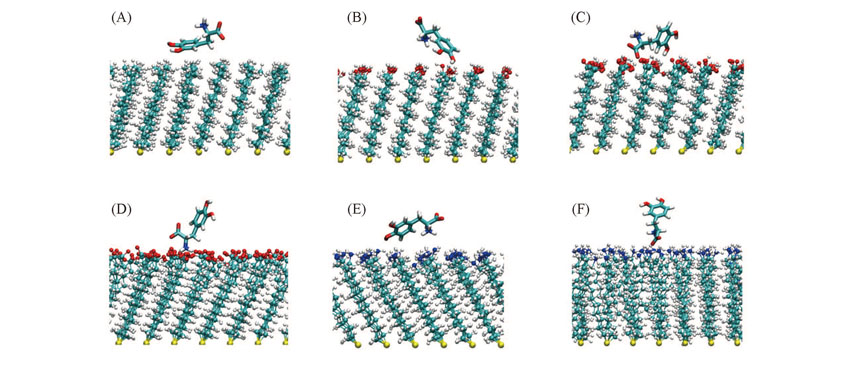
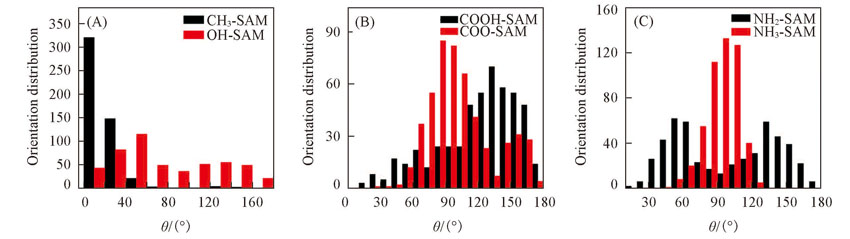
| [1] |
Waite J. H., Tanzer M. L., Science, 1981, 212(4498), 1038—1040
|
| [2] |
Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science, 2007, 318(5849), 426—430
|
| [3] |
Lu Q., Danner E., Waite J. H., Israelachvili J. N., Zeng H., Hwang D. S., J. R. Soc. Interface, 2013, 10(79), 20120759
|
| [4] |
Rego S. J., Vale A. C., Luz G. M., Mano J. F., Alves N. M., Langmuir, 2016, 32(2), 560—568
|
| [5] |
Tao K., Levin A., Adler-Abramovich L., Gazit E., Chem. Soc. Rev., 2016, 45(14), 3935—3953
|
| [6] |
Maity S., Nir S., Zada T., Reches M., Chem. Commun.(Camb), 2014, 50(76), 11154—11157
|
| [7] |
Lee B. P., Messersmith P. B., Israelachvili J. N., Waite J. H., Annu. Rev. Mater. Res., 2011, 41, 99—132
|
| [8] |
Lee H., Scherer N. F., Messersmith P. B., Proc. Natl. Acad. Sci. USA, 2006, 103(35), 12999—13003
|
| [9] |
Das P., Reches M., Biopolymers, 2015, 104(5), 480—494
|
| [10] |
Das P., Reches M., Nanoscale, 2016, 8(33), 15309—15316
|
| [11] |
Li Y., Qin M., Li Y., Cao Y., Wang W., Langmuir, 2014, 30(15), 4358—4366
|
| [12] |
Kinugawa S., Wang S., Taira S., Tsuge A., Kaneko D., Polymer Journal, 2016, 48(6), 715—721
|
| [13] |
Maier G. P., Rapp M. V., Waite J. H., Israelachvili J. N., Butler A., Science, 2015, 349(6248), 628—632
|
| [14] |
Li Y., Liu H., Wang T., Qin M., Cao Y., Wang W., Chem. Phys. Chem., 2016, 17, 1—5
|
| [15] |
Gao C. Y., Guo Y. Y., He J., Wu M., Liu Y., Chen Z. L., Cai W. S., Yang Y. L., Wang C., Feng X. Z., J. Mater. Chem., 2012, 22(21), 10763—10770
|
| [16] |
Yeh I. C., Lenhart J. L., Rinderspacher B. C., J. Phys. Chem. C, 2015, 119(14), 7721—7731
|
| [17] |
Qin Z., Buehler M., Appl. Phys. Lett., 2012, 101(8),083702-1—5
|
| [18] |
Peng C., Liu J., Zhao D., Zhou J., Langmuir, 2014, 30(38), 11401—11411
|
| [19] |
Peng C., Liu J., Xie Y., Zhou J., Phys. Chem. Chem. Phys., 2016, 18(15), 9979—9989
|
| [20] |
Liu J., Zhou J., Acta Biomater., 2016, 40, 23—30
|
| [21] |
Zhao D., Peng C., Zhou J., Phys. Chem. Chem. Phys., 2015, 17(2), 840—850
|
| [22] |
Liu J., Liao C., Zhou J., Langmuir, 2013, 29(36), 11366—11374
|
| [23] |
Liu J., Yu G., Zhou J., Chem. Eng. Sci., 2015, 121, 331—339
|
| [24] |
Xie Y., Liu M., Zhou J., Appl. Surf. Sci., 2012, 258(20), 8153—8159
|
| [25] |
Frazão N. F., Albuquerque E. L., Fulco U. L., Azevedo D. L., Mendonça G. L. F., Lima-Neto P., Caetano E. W. S., Santana J. V., Freire V. N., RSC Advances, 2012, 2(22), 8306—8322
|
| [26] |
Gfeller D., Michielin O., Zoete V., Nucleic Acids Res., 2013, 41(D1), D327—D332
|
| [27] |
MacKerell Jr A. D., Bashford D., Bellott M., Dunbrack Jr. R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., J. Phys. Chem. B, 1998, 102(18), 3586—3616
|
| [28] |
Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E., Bioinformatics, 2013, 29(7), 845—854
|
| [29] |
Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys., 1983, 79(2), 926—935
|
| [30] |
Bussi G., Donadio D., Parrinello M., J. Chem. Phys., 2007, 126(1),014101-1—8
|
| [31] |
Hess B., Bekker H., Berendsen H. J., Fraaije J. G., J. Comput. Chem., 1997, 18(12), 1463—1472
|
| [32] |
Darden T., York D., Pedersen L., J. Chem. Phys., 1993, 98(12), 10089—10092
|
| [33] |
Yeh I. C., Berkowitz M. L., J. Chem. Phys., 1999, 111(7), 3155—3162
|
| [34] |
Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 1996, 14(1), 33—38
|
| [35] |
Li L., Vorobyov I., Allen T. W., J. Phys. Chem. B, 2013, 117(40), 11906—11920
|
| [36] |
Souaille M., Roux B. T., Compt. Phys. Commun., 2001, 135(1), 40—57
|
| [37] |
He Z. J., Zhou J., Acta Chimica Sinica, 2011, 69(24), 2901—2907
|
|
(贺仲金, 周健.化学学报, 2011,69(24), 2901—2907)
|
| [38] |
Xu L., Hasin N., Shen M., He J., Xue Y., Zhou X., Perrett S., Song Y., Jones G. W., PLoS Comput. Biol., 2013, 9(1),e1002896-1—9
|
| [39] |
Zhou J., Zhang L., Leng Y., Tsao H. K., Sheng Y. J., Jiang S., J. Chem. Phys., 2006, 125(10),104905-1—104905-7
|
| [40] |
Levine Z. A., Rapp M. V., Wei W., Mullen R. G., Wu C., Zerze G. H., Mittal J., Waite J. H., Israelachvili J. N., Shea J. E., Proc. Natl. Acad. Sci. USA, 2016, 113(16), 4332—4337
|
| [41] |
Thyparambil A. A., Wei Y., Latour R. A., Langmuir, 2012, 28(13), 5687—5694
|
| [42] |
Zhang L. G., Xue Z. X., Zhang C., Yan H., Chem. J. Chinese Universities, 2016, 37(3), 505—512
|
|
(张鲁格, 薛泽旭, 张翀, 延辉.高等学校化学学报, 2016,37(3), 505—512)
|
| [43] |
Kang W. B., Xia Y., Wang J., Wang W., Acta Chimica Sinica, 2016, 74(8), 694—702
|
|
(康文斌, 夏耘, 王骏, 王炜.化学学报, 2016,74(8), 694—702)
|
 )
)