

高等学校化学学报 ›› 2014, Vol. 35 ›› Issue (10): 2138.doi: 10.7503/cjcu20140478
王秀军( ), 齐秋红, 陈丽
), 齐秋红, 陈丽
收稿日期:2014-05-23
出版日期:2014-10-10
发布日期:2014-09-15
作者简介:联系人简介: 王秀军, 男, 博士, 教授, 主要从事纳米材料的理论模拟研究. E-mail: 基金资助:
WANG Xiujun*(), QI Qiuhong, CHEN Li
Received:2014-05-23
Online:2014-10-10
Published:2014-09-15
Contact:
WANG Xiujun
E-mail:xjwangcn@scut.edu.cn
Supported by:摘要:
研究碳原子在TiO2(101)负载镍或铂原子上的吸附行为对于阐明积碳问题提供了一个热力学线索. 广义梯度近似密度泛函理论的PBE计算结果表明, 镍在TiO2表面最稳定构型的吸附能为347.16 kJ/mol, 铂对应的最稳定构型的吸附能为315.9 kJ/mol, 而且2种金属的最稳定构型均处于TiO2表面2个O2c原子之间的桥位. 吸附金属原子后, TiO2的态密度图中各电子峰向低能量方向移动, 体系趋于稳定. 从态密度图可知, 碳的p轨道与金属原子的d轨道发生叠加, 说明碳原子与金属原子成键, 从而使吸附后Ni或Pt与O原子之间的相互作用减弱. 碳原子吸附在Ni/TiO2(101)和Pt/TiO2(101)表面的最佳吸附结构的吸附能分别为474.19和570.08 kJ/mol, 说明TiO2负载铂催化剂在甲烷重整反应中抗积碳能力较强.
中图分类号:
TrendMD:
王秀军, 齐秋红, 陈丽. TiO2负载单金属镍(铂)催化剂在甲烷解离反应中抗积碳能力的理论研究. 高等学校化学学报, 2014, 35(10): 2138.
WANG Xiujun, QI Qiuhong, CHEN Li. Theoretical Studies on Suppression of Carbon Deposition over Titania Supported Monometallic Nickel(Platinum) Catalysts in Methane Dissociation†. Chem. J. Chinese Universities, 2014, 35(10): 2138.
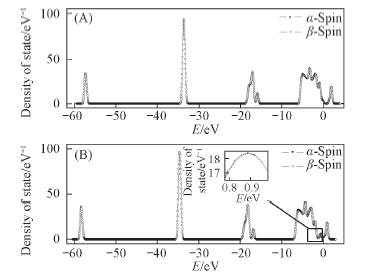
Fig.1 Density of states for the Pt(A) or Ni(B) atom adsorbed on TiO2(101) surfaceα-Spin: spin-up; β-spin: spin-down.
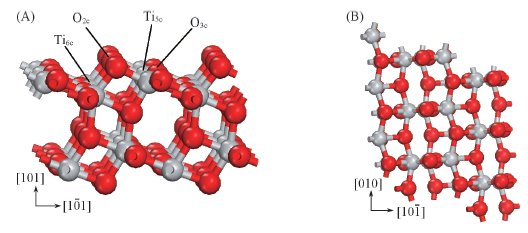
Fig.2 Side view(A) and top view(B) of the optimized structural models of the TiO2(101) clean surface
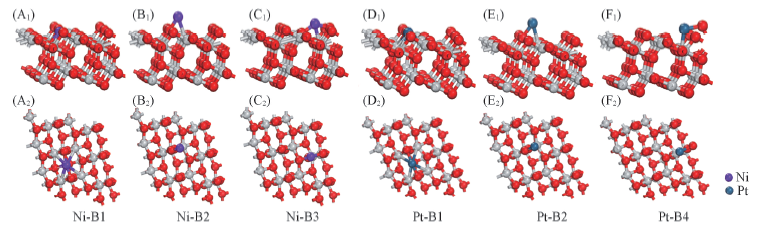
Fig.3 Side views(A1—F1) and top views(A2—F2) of the optimized configurations Ni and Pt adsorbed on the TiO2(101) surface
| Adsorption system | Eads/(kJ·mol-1) | ||||
|---|---|---|---|---|---|
| Ni-B1 | 347.16 | 0.1904 | 0.2056 | 0.2774 | 0.2658 |
| Pt-B1 | 315.90 | 0.2059(0.204)b | 0.2778(0.278)b | 0.2826(0.279)b | |
| Ni-B3 | 311.61 | 0.1844 | 0.1862 | 0.2315 | |
| Pt-B4 | 309.60 | 0.2015 | 0.2353 | ||
| Ni-B2 | 244.22 | 0.1765 | 0.2419 | ||
| Pt-B2 | 235.35 | 0.1972 | 0.2498 |
Table 1 Optimazated chemisorptions energies and structural parameters of Ni(Pt) atom adsorbed on anatase TiO2(101) surface
| Adsorption system | Eads/(kJ·mol-1) | ||||
|---|---|---|---|---|---|
| Ni-B1 | 347.16 | 0.1904 | 0.2056 | 0.2774 | 0.2658 |
| Pt-B1 | 315.90 | 0.2059(0.204)b | 0.2778(0.278)b | 0.2826(0.279)b | |
| Ni-B3 | 311.61 | 0.1844 | 0.1862 | 0.2315 | |
| Pt-B4 | 309.60 | 0.2015 | 0.2353 | ||
| Ni-B2 | 244.22 | 0.1765 | 0.2419 | ||
| Pt-B2 | 235.35 | 0.1972 | 0.2498 |
| Adsorption site | qNi(Pt)/e | /e | /e | /e | /e |
|---|---|---|---|---|---|
| Ni-B1 | 0.49 | -0.66 | -0.70 | 1.30 | 1.25 |
| Ni-B2 | 0.43 | -0.65 | 1.06 | ||
| Ni-B3 | 0.46 | -0.66 | -0.72 | 1.11 | |
| Pt-B1 | 0.18 | -0.64 | 1.30 | 1.24 | |
| Pt-B2 | 0.18 | -0.62 | 1.17 | ||
| Pt-B4 | 0.21 | -0.57 | 1.09 |
Table 2 Mulliken charge population of Ni(Pt) atom adsorbed on TiO2(101) surface
| Adsorption site | qNi(Pt)/e | /e | /e | /e | /e |
|---|---|---|---|---|---|
| Ni-B1 | 0.49 | -0.66 | -0.70 | 1.30 | 1.25 |
| Ni-B2 | 0.43 | -0.65 | 1.06 | ||
| Ni-B3 | 0.46 | -0.66 | -0.72 | 1.11 | |
| Pt-B1 | 0.18 | -0.64 | 1.30 | 1.24 | |
| Pt-B2 | 0.18 | -0.62 | 1.17 | ||
| Pt-B4 | 0.21 | -0.57 | 1.09 |
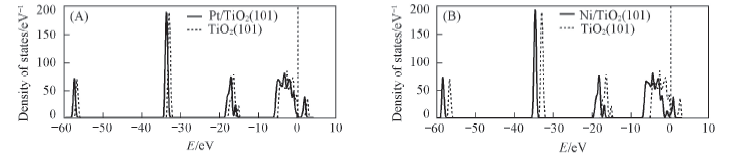
Fig.4 Density of states for the TiO2(101) surface and the most stable structural of Pt/TiO2(101) surface(A) and Ni/TiO2(101) surface(B) The vertical dot lines at the energy zero represent the Fermi level.
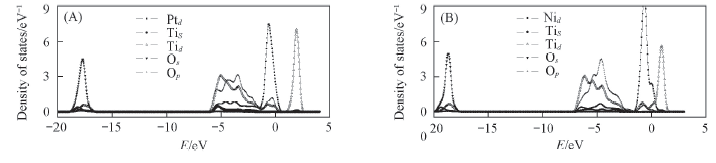
Fig.5 Partial density of states for the most stable structural of Pt/TiO2(101)(A) and Ni/TiO2(101)(B)
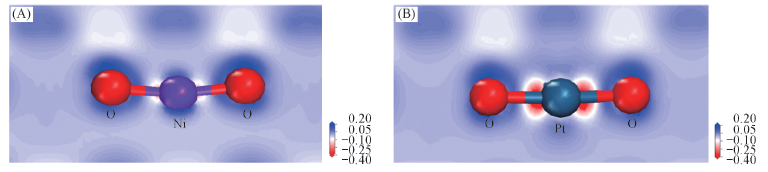
Fig.6 Charge difference density contour maps of the most stable structural of Ni(Pt) adsorbed on TiO2(101) surface (A) Ni-B1; (B) Pt-B1.
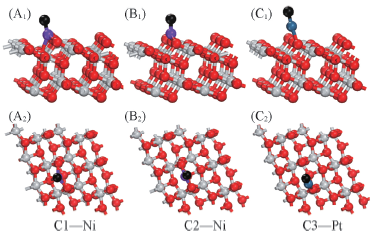
Fig.7 Side views(A1—C1) and top views(A2—C2)of the optimized configuration about the carbon atoms on Ni(Pt)/TiO2(101) surface Black balls indicate C atoms.
| Adsorption site | Eads/(kJ·mol-1) | RC—Ni(Pt)/nm | qC/e | qNi(Pt)/e | ||||
|---|---|---|---|---|---|---|---|---|
| C1—Ni | 474.19 | 0.1623 | 0.1986* | 0.2236 | -0.16 | 0.60 | -0.69 | -0.72 |
| C2—Ni | 462.63 | 0.1621 | 0.1939* | -0.15 | 0.56 | -0.71 | ||
| C3—Pt | 570.08 | 0.1754 | 0.2201 | -0.10 | 0.10 | -0.71 |
Table 3 Adsorption energies(Eads), geometric parameters of different configurations about carbon atom adsorbed on Ni(Pt)/TiO2(101) surface(R) and the mulliken population for C atom adsorbed on Ni(Pt)/TiO2(101) surface(q)
| Adsorption site | Eads/(kJ·mol-1) | RC—Ni(Pt)/nm | qC/e | qNi(Pt)/e | ||||
|---|---|---|---|---|---|---|---|---|
| C1—Ni | 474.19 | 0.1623 | 0.1986* | 0.2236 | -0.16 | 0.60 | -0.69 | -0.72 |
| C2—Ni | 462.63 | 0.1621 | 0.1939* | -0.15 | 0.56 | -0.71 | ||
| C3—Pt | 570.08 | 0.1754 | 0.2201 | -0.10 | 0.10 | -0.71 |
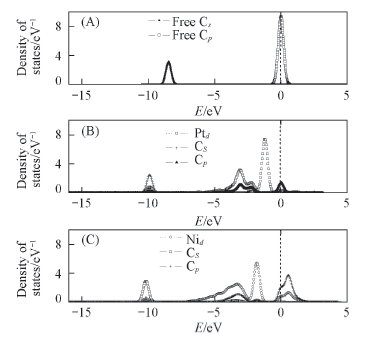
Fig.8 Partial density of the states for the most stable structural of C adsorbed on Ni(Pt)/TiO2(101) surface (A) Free C; (B) C on the Pt/TiO2(101) surface; (C) C adsorbed on the Ni/TiO2(101) surface.
| [1] | Luntz A. C., Bethune D. S., J. Chem. Phys., 1989, 90(2), 1274—1280 |
| [2] | San Jose-Alonso D., Illan-Gomez M. J., Roman-Martinez M. C., Int. J. Hydrog. Energy,2013, 38(5), 2230—2239 |
| [3] | Lv X. Y., Chen J. F., Tan Y. S., Zhang Y., Catal. Commun., 2012, 20(5), 6—11 |
| [4] | Solymosi F., Erdohelyi A., Cserenyi J., Felvegi A., J. Catal., 1994, 147(1), 272—278 |
| [5] | Mateos-Pedrero C., Gonzalez-Carrazan S. R., Soria M. A., Ruiz P., Catal. Today,2013, 203(30), 158—162 |
| [6] | Zhang Z. L., Verykios X. E., App. Catal. A,1996, 138(1), 109—133 |
| [7] | Nagaoka K., Seshan K., Aika K., Lercher J. A., J. Catal., 2001, 197(1), 34—42 |
| [8] | Bitter J., Seshan K., Lercher J., J. Catal., 1998, 176(1), 93—101 |
| [9] | Li G. S., Li L. P., Boerio-Goates J., Woodfield B. F., J. Am. Chem. Soc., 2005, 127(24), 8659—8666 |
| [10] | Lazzeri M., Vittadini A., Selloni A., Phys. Rev. B,2002, 65(11), 119901 |
| [11] | Herman G. S., Dohnalek Z., Ruzycki N., Diebold U., J. Phys. Chem. B,2003, 107(12), 2788—2795 |
| [12] | Hengerer R., Bolliger B., Erbudak M., Gratzel M., Surf. Sci., 2000, 460(1/3), 162—169 |
| [13] | Lazzeri M., VittadiniA., Selloni A., Phys. Rev. B,2001, 63(15), 155409—155418 |
| [14] | Wang F. C., Wan H. L., Tsai K. R., Wang S. J., Xu F. C., Catal. Lett., 1992, 12(1/3), 319—325 |
| [15] | Stakheev A. Y., Gololobov A. M., Beck I. E., Bragina G. O., Zaikovsky V. I., Ayupov A. B., Telegina N. S., Bukhtiyarov V. I., Russ. Chem. Bull., 2010, 59(9), 1713—1719 |
| [16] | Zou J. J., He H., Cui L., Du H. Y., Int. J. Hydrog. Energy,2007, 32(12), 1762—1770 |
| [17] | Bitter J. H., Seshan K., Lercher J. A., J. Catal., 1999, 183(2), 336—343 |
| [18] | Li C. L., Chen W., Yuan J., Shangguan W. F., Acta Phys. Chim. Sinica,2012, 28(2), 450—456 |
| (李曹龙, 陈威, 袁坚, 上官文峰. 物理化学学报, 2012, 28(2), 450—456) | |
| [19] | Bradford M. C., Vannice M. A., Appl. Catal. A,1996, 142(1), 73—96 |
| [20] | Bradford M. C., Vannice M. A., Appl. Catal. A,1996, 142(1), 97—122 |
| [21] | Yan Q. G., Weng W.Z., Wan H. L., Toghiani H., Toghiani R. K., Pittman C. U., Appl. Catal. A,2003, 239(1/2), 43—58 |
| [22] | Takanabe K., Nagaoka K., Nariai K., Aika K., J. Catal., 2005, 232(2), 268—275 |
| [23] | Wanbayor R., Ruangpornvisuti V., Appl. Surf. Sci., 2012, 258(7), 3298—3301 |
| [24] | Zhou Y., Muhich C. L., Neltner B. T., Weimer A. W., Musgrave C. B., J. Phys. Chem. C,2012, 116(22), 12114—12123 |
| [25] | Celik V., Unal H., Mete E., Ellialtioglu S., Phys. Rev. B,2010, 82(20), 205113—205125 |
| [26] | Zhang R. G., Song L. Z., Wang Y. H., Appl. Surf. Sci., 2012, 258(18), 7154—7160 |
| [27] | He P. L., Mao Y. L., Sun L. Z., Zhong J. X., J. Comput. Theor. Nanosci., 2010, 7(10), 2063—2067 |
| [28] | Escamilla-Roa E., Timon V., Hernandez-Laguna A., Comput. Theor. Chem., 2012, 981(1), 59—67 |
| [29] | Yang Z. X., He B. L., Lu Z. S., Hermansson K. T., J. Phys. Chem. C,2010, 114(10), 4486—4494 |
| [30] | Cheng D. J., Lan J. H., Cao D. P., Wang W. C., Appl. Catal. B,2010, 106(3/4), 510—519 |
| [31] | Delley B., J. Phys. Chem., 1996, 100(15), 6107—6110 |
| [32] | Delley B., J. Chem. Phys., 1990, 92(1), 508—517 |
| [33] | Delley B., Phys. Rev. B, 2002, 66(15), 155125—155134 |
| [34] | Perdew J. P., Chevary J. A., Vosko S. H., Jackson K. A., Pederson M. R., Singh D. J., Fiolhais C., Phys. Rev. B,1992, 46(11), 6671—6687 |
| [35] | Ernzerhof M., Scuseria G. E., J. Chem. Phys., 1999, 110(11), 5029—5036 |
| [36] | Iwaszuk A., Mulheran P. A., Nolan M., J. Mater. Chem. A,2013, 1(7), 2515—2525 |
| [37] | Burdett J. K., Hughbanks T., Miller G. J., Richardson J. W. Jr, Smith J. V., J. Am. Chem. Soc., 1987, 109(12), 3639—3646 |
| [38] | Goldwasser M. R., Rivas M. E., Pietri E., Perez-Zurita M. J., Cubeiro M. L., Gingembre L., Leclercq L., Leclercq G., Appl. Catal. A,2003, 255(1), 45—57 |
| [39] | Ding K. N., Zhang Y. F., Li Y., Li J. J., J. Fuzhou. Univ. Nat. Sci. Ed., 2005, 33(4), 528—532 |
| (丁开宁, 章永凡, 李奕, 李俊籛. 福州大学学报自然科学版, 2005, 33(4), 528—532) | |
| [40] | Han Y., Liu C. J., Ge Q. F., J. Phys. Chem. B,2006, 110(14), 7463—7472 |
| [41] | Li S. R., Lu X. Q., Guo W. Y., Zhu H. Y., Li M., Zhao L. M., Li Y., Shan H. H., J. Organomet. Chem., 2012, 704(1), 38—48 |
| [42] | Chen Y. F., Zhang M. H., Jiang H. X., Mol. Catal.(China), 2007, 21(4), 351—355 |
| (陈毅飞, 张敏华, 姜浩锡. 分子催化, 2007, 21(4), 351—355) | |
| [43] | Zhao Y., Teng B. T., Wen X. D., Zhao Y., Zhao L. H., Luo M. F., Catal. Commun., 2012, 27(5), 63—68 |
| [44] | Zhao Y., Teng B. T., Yang Z. X., Zhao Y., Zhao L. H., Luo M., J. Phys. Chem. C,2011, 115(33), 16461—16466 |
| [45] | Zhang W. D., Liu B. S., Zhu C., Tian Y. L., Appl. Catal. A,2005, 292(18), 138—143 |
| [46] | Fan C., Zhu Y. A., Zhou X. G., Chem. Re. Eng. Technol., 2012, 28(2), 117—121 |
| [47] | Xu Y., Ruban A. V., Mavrikakis M., J. Am. Chem. Soc., 2004, 126(14), 4717—4725 |
| [48] | Greeley J., Norskov J. K., Surf. Sci., 2005, 592(1/3), 104—111 |
| [49] | Tomishige K., Kanazawa S., Suzuki K., Asadullah M., Sato M., Ikushima K., Kunimori K., Appl. Catal. A,2002, 233(1/2), 35—44 |
| [50] | Wang S. B., Lu G. Q., Millar G. J., Energ. Fuel,1996, 10(4), 896—904 |
| [1] | 任诗杰, 谯思聪, 刘崇静, 张文华, 宋礼. 铂单原子催化剂同步辐射X射线吸收谱的研究进展[J]. 高等学校化学学报, 2022, 43(9): 20220466. |
| [2] | 韦春洪, 蒋倩, 王盼盼, 江成发, 刘岳峰. 贵金属Pt促进Co基费托合成催化剂的原子尺度结构分析[J]. 高等学校化学学报, 2022, 43(8): 20220074. |
| [3] | 姜宏斌, 代文臣, 张娆, 徐晓晨, 陈捷, 杨光, 杨凤林. Co3O4/UiO-66@α-Al2O3陶瓷膜对VOCs废气的分离催化性能[J]. 高等学校化学学报, 2022, 43(6): 20220025. |
| [4] | 戴卫, 侯华, 王宝山. 七氟异丁腈负离子结构与反应活性的理论研究[J]. 高等学校化学学报, 2022, 43(6): 20220044. |
| [5] | 郝宏蕾, 孟繁雨, 李若钰, 李迎秋, 贾明君, 张文祥, 袁晓玲. 生物质基氮掺杂多孔炭材料的制备及对水中亚甲基蓝的吸附性能[J]. 高等学校化学学报, 2022, 43(6): 20220055. |
| [6] | 王广琦, 毕艺洋, 王嘉博, 石洪飞, 刘群, 张钰. 非贵金属三元复合Ni(PO3)2-Ni2P/CdS NPs异质结的构建及可见光高效催化产氢性能[J]. 高等学校化学学报, 2022, 43(6): 20220050. |
| [7] | 胡慧敏, 崔静, 刘丹丹, 宋佳欣, 张宁, 范晓强, 赵震, 孔莲, 肖霞, 解则安. 过渡金属修饰对Pt/M-DMSN催化剂丙烷脱氢性能的影响[J]. 高等学校化学学报, 2022, 43(4): 20210815. |
| [8] | 王红宁, 黄丽, 清江, 马腾洲, 蒋伟, 黄维秋, 陈若愚. 香蒲基生物炭的活化及对VOCs吸附的应用[J]. 高等学校化学学报, 2022, 43(4): 20210824. |
| [9] | 陈潇禄, 袁珍闫, 仲迎春, 任浩. 机械球磨制备三苯胺基PAF-106s及C2烃吸附性质[J]. 高等学校化学学报, 2022, 43(3): 20210771. |
| [10] | 孟祥龙, 杨歌, 郭海玲, 刘晨光, 柴永明, 王纯正, 郭永梅. 纳米分子筛的合成及硫化氢吸附性能[J]. 高等学校化学学报, 2022, 43(3): 20210687. |
| [11] | 李晓辉, 魏爱佳, 穆金萍, 何蕊, 张利辉, 王军, 刘振法. 磷酸钐包覆对高电压镍锰酸锂正极材料电化学性能的影响[J]. 高等学校化学学报, 2022, 43(2): 20210546. |
| [12] | 靳科研, 白璞, 李小龙, 张佳楠, 闫文付. 新型Mg-Al吸附剂去除压水堆核电厂废水中高浓度硼[J]. 高等学校化学学报, 2022, 43(2): 20210516. |
| [13] | 郭彪, 赵晨灿, 刘芯辛, 于洲, 周丽景, 袁宏明, 赵震. 表面水热碳层对磁性NiFe2O4八面体光催化活性的影响[J]. 高等学校化学学报, 2022, 43(11): 20220472. |
| [14] | 谭乐见, 仲宣树, 王锦, 刘宗建, 张爱英, 叶霖, 冯增国. β-环糊精的低临界溶解温度现象及其在有序纳米孔道片晶制备中的应用[J]. 高等学校化学学报, 2022, 43(11): 20220405. |
| [15] | 郑美琪, 毛方琪, 孔祥贵, 段雪. 类水滑石材料在核废水处理领域的应用[J]. 高等学校化学学报, 2022, 43(10): 20220456. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||