

Chem. J. Chinese Universities ›› 2016, Vol. 37 ›› Issue (4): 693.doi: 10.7503/cjcu20150844
• Physical Chemistry • Previous Articles Next Articles
JIANG Junhui, XIA Shengjie, NI Zheming( ), Zhang Lianyang
), Zhang Lianyang
Received:2015-11-06
Online:2016-04-10
Published:2016-03-17
Supported by:CLC Number:
TrendMD:
JIANG Junhui, XIA Shengjie, NI Zheming, Zhang Lianyang. Adsorption and Selective Hydrogenation Mechanism of Crotonaldehyde on AuSurface[J]. Chem. J. Chinese Universities, 2016, 37(4): 693.
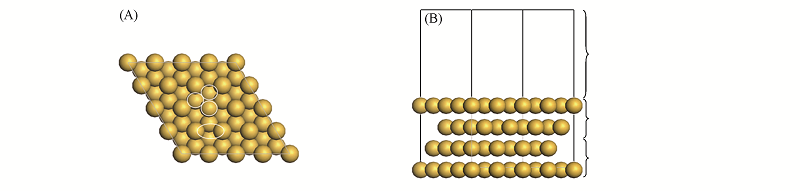
Fig.1 Top(A) and side(B) views of Au(111) surface models(4×4)^The four representative surface sites(Top, Bri, Hcp and Fcc site) are indicated in the top view.
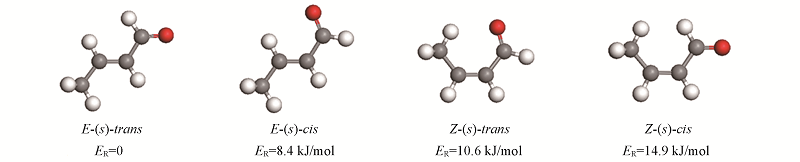
Fig.2 Configuration and relative energy of four isomers of CAL
| Initial adsorption site | Final adsorption site | Eads/(kJ·mol-1) | Initial adsorption site | Final adsorption site | Eads/(kJ·mol-1) |
|---|---|---|---|---|---|
| O | C | ||||
| Top | Top | -85.1 | Top-Top | Bri-Top | -84.5 |
| Bri | Bri | -86.8 | Top-Bri | Bri-Bri | -84.5 |
| Hcp | Hcp | -84.1 | Top-Hcp | Top-Hcp | -84.2 |
| Fcc | Fcc | -84.9 | Top-Fcc | Top-Fcc | -84.3 |
| C | Bri-Top | Bri-Top | -84.5 | ||
| Top | Top | -93.7 | Bri-Bri | Bri-Bri | -84.6 |
| Bri | Bri | -90.5 | Bri-Hcp | Bri-Top | -84.0 |
| Hcp | Hcp | -85.0 | Bri-Fcc | Bri-Fcc | -83.8 |
| Fcc | Fcc | -86.7 | Hcp-Top | Hcp-Top | -84.7 |
| C | Hcp-Bri | Hcp-Bri | -84.5 | ||
| Top | Top | -79.7 | Hcp-Hcp | Bri-Fcc | -83.5 |
| Bri | Bri | -80.4 | Hcp-Fcc | Hcp-Fcc | -84.0 |
| Hcp | Hcp | -81.8 | Fcc-Top | Fcc-Top | -84.4 |
| Fcc | Fcc | -82.6 | Fcc-Bri | Fcc-Bri | -83.5 |
| Fcc-Hcp | Bri-Bri | -84.6 | |||
| Fcc-Fcc | Fcc-Fcc | -83.7 |
Table 1 Adsorption energies of CAL on Au(111) surface
| Initial adsorption site | Final adsorption site | Eads/(kJ·mol-1) | Initial adsorption site | Final adsorption site | Eads/(kJ·mol-1) |
|---|---|---|---|---|---|
| O | C | ||||
| Top | Top | -85.1 | Top-Top | Bri-Top | -84.5 |
| Bri | Bri | -86.8 | Top-Bri | Bri-Bri | -84.5 |
| Hcp | Hcp | -84.1 | Top-Hcp | Top-Hcp | -84.2 |
| Fcc | Fcc | -84.9 | Top-Fcc | Top-Fcc | -84.3 |
| C | Bri-Top | Bri-Top | -84.5 | ||
| Top | Top | -93.7 | Bri-Bri | Bri-Bri | -84.6 |
| Bri | Bri | -90.5 | Bri-Hcp | Bri-Top | -84.0 |
| Hcp | Hcp | -85.0 | Bri-Fcc | Bri-Fcc | -83.8 |
| Fcc | Fcc | -86.7 | Hcp-Top | Hcp-Top | -84.7 |
| C | Hcp-Bri | Hcp-Bri | -84.5 | ||
| Top | Top | -79.7 | Hcp-Hcp | Bri-Fcc | -83.5 |
| Bri | Bri | -80.4 | Hcp-Fcc | Hcp-Fcc | -84.0 |
| Hcp | Hcp | -81.8 | Fcc-Top | Fcc-Top | -84.4 |
| Fcc | Fcc | -82.6 | Fcc-Bri | Fcc-Bri | -83.5 |
| Fcc-Hcp | Bri-Bri | -84.6 | |||
| Fcc-Fcc | Fcc-Fcc | -83.7 |
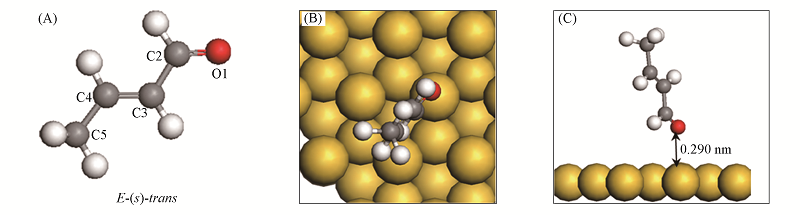
Fig.3 Structure of CAL(A) and its most stable configuration(B, C) on Au(111) surface^ (B) Top view; (C) side view.
| Species | Charge/e | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O1 | C2 | C3 | C4 | C5 | H6 | H7 | H8 | H9 | H10 | H11 | Tol | |
| CAL | -0.378 | 0.282 | -0.050 | 0.010 | -0.135 | -0.009 | 0.042 | 0.044 | 0.055 | 0.069 | 0.070 | 0.000 |
| CAL/Au(111) | -0.355 | 0.232 | -0.086 | -0.033 | -0.281 | 0.032 | 0.099 | 0.100 | 0.099 | 0.118 | 0.120 | 0.045 |
Table 2 Mulliken charges of CAL at advantage adsorption site on Au(111) surface
| Species | Charge/e | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O1 | C2 | C3 | C4 | C5 | H6 | H7 | H8 | H9 | H10 | H11 | Tol | |
| CAL | -0.378 | 0.282 | -0.050 | 0.010 | -0.135 | -0.009 | 0.042 | 0.044 | 0.055 | 0.069 | 0.070 | 0.000 |
| CAL/Au(111) | -0.355 | 0.232 | -0.086 | -0.033 | -0.281 | 0.032 | 0.099 | 0.100 | 0.099 | 0.118 | 0.120 | 0.045 |
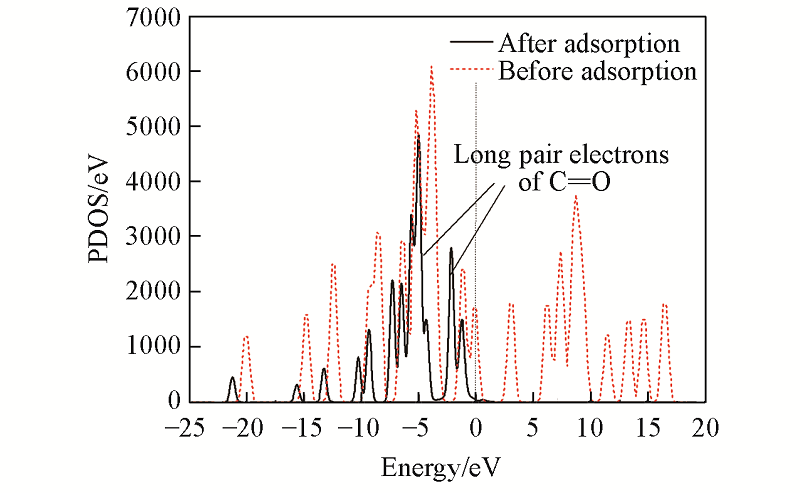
Fig.4 p oribital partial density of states of CAL before and after adsorption
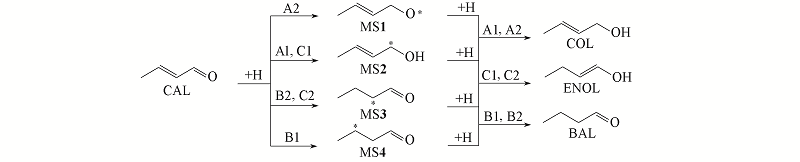
Fig.5 Different reaction mechanisms for the partial hydrogenation of CAL
| Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) | Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) |
|---|---|---|---|---|---|---|---|
| A1 | CAL*+H*→MS2*+* | 35.1 | -43.8 | C1 | CAL*+H*→MS2*+* | 35.1 | -43.8 |
| MS2*+H*→COL*+* | 150.7 | -47.5 | MS2*+H*→ENOL*+* | 312.2 | -89.6 | ||
| A2 | CAL*+H*→MS1*+* | 70.3 | -10.2 | C2 | CAL*+H*→MS3*+* | 288.1 | -0.4 |
| MS1*+H*→COL*+* | 28.4 | -81.0 | MS3*+H*→ENOL*+* | 42.6 | -133.0 | ||
| B1 | CAL*+H*→MS4*+* | 240.2 | -32.5 | CAL+*→CAL* | -93.7 | ||
| MS4*+H*→BAL*+* | 301.7 | -124.7 | COL*→COL +* | 51.6 | |||
| B2 | CAL*+H*→MS3*+* | 288.1 | -0.4 | ENOL*→ENOL +* | 56.2 | ||
| MS3*+H*→BAL*+* | 220.3 | -156.9 | BAL*→BAL +* | 60.9 |
Table 3 Activation energy(Ea) and reaction energy(ΔE) of main elementary reactions for the partial hydrogenation of CAL on Au(111) surface
| Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) | Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) |
|---|---|---|---|---|---|---|---|
| A1 | CAL*+H*→MS2*+* | 35.1 | -43.8 | C1 | CAL*+H*→MS2*+* | 35.1 | -43.8 |
| MS2*+H*→COL*+* | 150.7 | -47.5 | MS2*+H*→ENOL*+* | 312.2 | -89.6 | ||
| A2 | CAL*+H*→MS1*+* | 70.3 | -10.2 | C2 | CAL*+H*→MS3*+* | 288.1 | -0.4 |
| MS1*+H*→COL*+* | 28.4 | -81.0 | MS3*+H*→ENOL*+* | 42.6 | -133.0 | ||
| B1 | CAL*+H*→MS4*+* | 240.2 | -32.5 | CAL+*→CAL* | -93.7 | ||
| MS4*+H*→BAL*+* | 301.7 | -124.7 | COL*→COL +* | 51.6 | |||
| B2 | CAL*+H*→MS3*+* | 288.1 | -0.4 | ENOL*→ENOL +* | 56.2 | ||
| MS3*+H*→BAL*+* | 220.3 | -156.9 | BAL*→BAL +* | 60.9 |
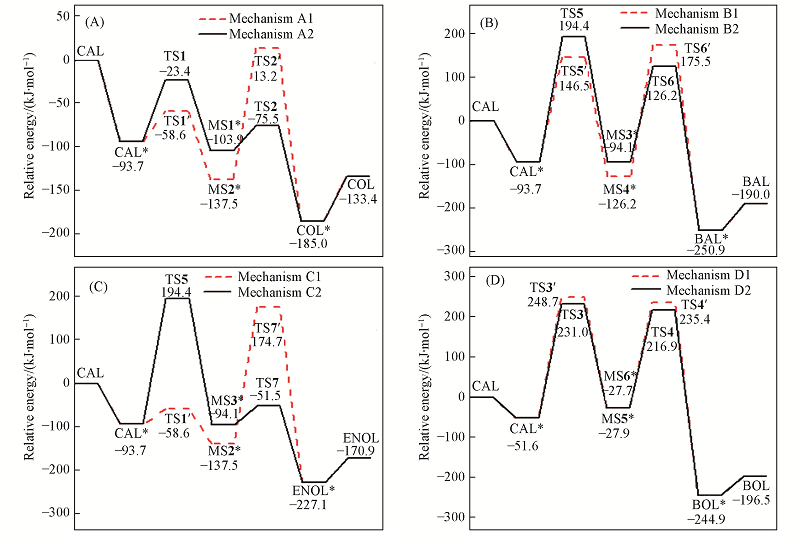
Fig.6 Sketch for potential relative energy of reaction mechanisms on Au(111) surface^ (A) Mechanism A; (B) mechanism B; (C) mechanism C; (D) mechanism D.
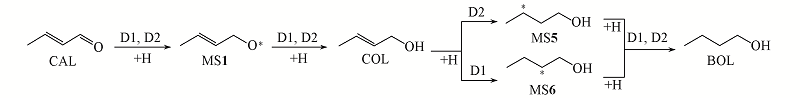
Fig.7 Different reaction mechanisms for the selective hydrogenation of CAL
| Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) | Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) |
|---|---|---|---|---|---|---|---|
| D1 | CAL+*→CAL* | -93.7 | D2 | CAL*+H*→MS1*+* | 70.3 | -10.2 | |
| CAL*+H*→MS1*+* | 70.3 | -10.2 | MS1*+H*→COL*+* | 28.4 | -81.0 | ||
| MS1*+H*→COL*+* | 28.4 | -81.0 | COL*+H*→MS5*+* | 282.6 | 23.7 | ||
| COL*+H*→MS6*+* | 300.3 | 23.9 | MS5*+H*→BOL*+* | 244.8 | -217.0 | ||
| MS6*+H*→BOL*+* | 263.1 | -217.2 | BOL*→BOL+* | 48.4 |
Table 4 Activation energy(Ea) and reaction energy(ΔE) of main elementary reactions for the full hydrogenation of CAL on Au(111) surface
| Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) | Mechanism | Reaction | Ea/ (kJ·mol-1) | ΔE/ (kJ·mol-1) |
|---|---|---|---|---|---|---|---|
| D1 | CAL+*→CAL* | -93.7 | D2 | CAL*+H*→MS1*+* | 70.3 | -10.2 | |
| CAL*+H*→MS1*+* | 70.3 | -10.2 | MS1*+H*→COL*+* | 28.4 | -81.0 | ||
| MS1*+H*→COL*+* | 28.4 | -81.0 | COL*+H*→MS5*+* | 282.6 | 23.7 | ||
| COL*+H*→MS6*+* | 300.3 | 23.9 | MS5*+H*→BOL*+* | 244.8 | -217.0 | ||
| MS6*+H*→BOL*+* | 263.1 | -217.2 | BOL*→BOL+* | 48.4 |
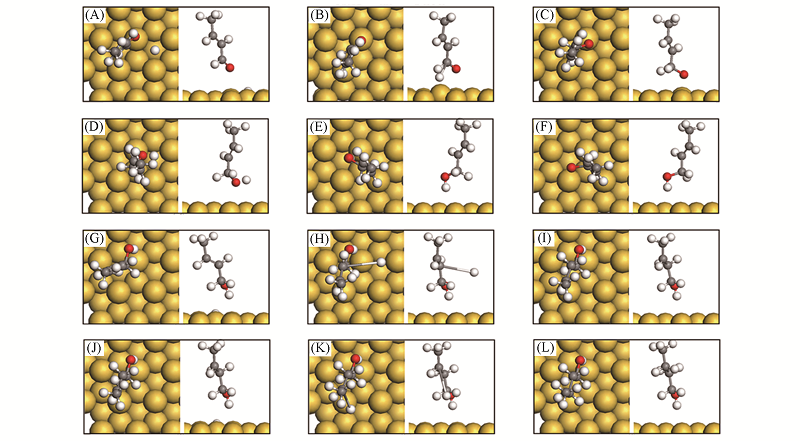
Fig.8 Structure change for reactants of mechanisms A2 and D2 on Au(111) surface^(A)—(C) CAL*+H*→MS1*+*; (D)—(F) MS1*+H*→COL*+*; (G)—(I) COL*+H*→MS5*+*; (J)—(L) MS5*+H*→BOL*+*. (A) IS1; (B) TS1; (C) MS1; (D) IS2; (E) TS2; (F) COL; (G) IS3; (H) TS3; (I) MS5; (J) IS4; (K) TS4; (L) BOL.
| [1] | Li X. H., Zheng W. L., Pan H. Y., J. Catal., 2013, 300, 9—19 |
| [2] | Galletti A. M. R., Toniolo L., Antonetti C., Evangelisti C., Forte C., Appl. Catal. A-Gen., 2012, 447, 49—59 |
| [3] | Hong X., Li B., Wang Y. J., Lu J. Q., Hu G. S., Luo M. F., Appl. Surf. Sci., 2013, 270, 388—394 |
| [4] | Nholler H., Lin W. M., J. Catal., 1984, 85, 25—30 |
| [5] | Fang C., Chen Y. J., Mao H., Zhao J., Jiang Y. F., Zhao S. L., Ma J., Chem. J. Chinese Universities, 2015, 36(1), 124—130 |
| (方超, 陈亚君, 毛卉, 赵俊, 蒋云福, 赵仕林, 马骏. 高等学校化学学报, 2015, 36(1), 124—130) | |
| [6] | Hisahiro H., Tomomi N., Takashi H., Toshio S., Green Chem., 2011, 13, 1133—1137 |
| [7] | Rodrigues E. L., Bueno J. M. C., Appl. Catal. A-Gen., 2004, 257, 201—211 |
| [8] | Tian Z. B., Li Q. Y., Li Y., Ai S. Y., Catal. Commun., 2015, 61, 97—101 |
| [9] | Aoun M., Benamar A., Chater M., Chinese J. Catal., 2011, 32, 1185—1190(Aoun M., Benamar A., Chater M., 催化学报, 2011,32, 1185—1190) |
| [10] | Bailie J.E., Hutching G. J.,Chem. Commun., 1999, (21), 2151—2152 |
| [11] | Bailie J. E., Abdullah H. A., Anderson J. A., Rochester C. H., Richardson N. V., Hodge N., Zhang J. G., Burrows A., Kiely C. J., Hutchings G. J., Phys. Chem. Chem. Phys., 2001, 3, 4113—4121 |
| [12] | Mohr C., Hofmeister H., Radnik J., Claus P., J. Am. Chem. Soc., 2003, 125, 1905—1911 |
| [13] | Zhao J., Ni J., Xu J. H., Xu J. T., Cen J., Li X. N., Catal. Commun., 2014, 54, 72—76 |
| [14] | Pan W., Ma W. G., Yang X. D., Zheng J. Y., Song B. Q., Niu Y. Z., Gu J., Hu D. B., Yang Q., Zhu H. J., Chem. J. Chinese Universities, 2015, 36(2), 325—329 |
| (潘威, 马文广, 杨晓东, 郑昀晔, 宋碧清, 牛永志, 古吉, 胡栋宝, 杨芹, 朱华结. 高等学校化学学报, 2015, 36(2), 325—329) | |
| [15] | Gholizadeh R., Yu Y. X., Appl. Surf. Sci., 2015, 357, 1187—1195 |
| [16] | Liu T. T., Lu X., Zhang M. T., Chem. Res. Chinese Universities, 2014, 30(4), 656—660 |
| [17] | Stefanov B. I., Topalian Z., Granqvist C. G., Osterlund L., J. Mol. Catal. A-Chem., 2014, 381, 77—88 |
| [18] | Shi W., Zhang L. Y., Ni Z. M., Xiao X. C., Xia S. J., RSC Adv., 2014, 4, 27003—27012 |
| [19] | Cao X. M., Burch R., Hardacre C., Hu P., J. Phys. Chem. C, 2011, 115, 19819—19827 |
| [20] | Yu Y. X.,ACS Appl. Mater. Interfaces, 2014, 6, 16267—16275 |
| [21] | Yu Y. X., J. Mater. Chem. A, 2014, 2, 8910—8917 |
| [22] | Ge Q., Jenkins S. J., King D. A., Chem. Phys. Lett., 2000, 327, 125—130 |
| [23] | Ni Z. M., Shi W., Xia M. Y., Xue J. L., Chem. J. Chinese Universities, 2013, 34(10), 2353—2362 |
| (倪哲明, 施炜, 夏明玉, 薛继龙. 高等学校化学学报, 2013, 34(10), 2353—2362) | |
| [24] | Zhao X. D., Song L. Z., Fu J., Tang P., Liu F., Surf. Sci., 2011, 605, 1005—1015 |
| [25] | Zhang L.Y., Jiang J.H., Shi W., Xia S. J., Ni Z. M., Xiao X. C., RSC Adv., 2015, 5, 34319—34326 |
| [26] | Haubrich J., Loffreda D., Delbecq F., Sautet P., Krupski K., Becker C., Wandeltt K., J. Phys. Chem. C, 2009, 113, 13947—13967 |
| [27] | Xiao X. C., Shi W., Ni Z. M., Acta Phys-Chim. Sinica, 2014, 30, 1456—1464 |
| (肖雪春, 施炜, 倪哲明. 物理化学学报, 2014, 30, 1456—1464) | |
| [28] | Delbecq D., Sautet P., J. Catal., 1995, 152, 217—236 |
| [29] | Xie Y., Yu H. T., Chem. Res. Chinese Universities, 2014, 30(5), 794—799 |
| [30] | Mullken R. S., J. Chem. Phys., 1955, 23, 1833—1840 |
| [31] | Loffreda D., Delbecq F., Vigne F., Sautet P., J. Am. Chem. Soc., 2006, 128, 1316—1323 |
| [32] | Peter C., Appl. Catal. A-Gen., 2005, 291, 222—229 |
| [1] | HE Hongrui, XIA Wensheng, ZHANG Qinghong, WAN Huilin. Density-functional Theoretical Study on the Interaction of Indium Oxyhydroxide Clusters with Carbon Dioxide and Methane [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220196. |
| [2] | JIANG Hongbin, DAI Wenchen, ZHANG Rao, XU Xiaochen, CHEN Jie, YANG Guang, YANG Fenglin. Research on Co3O4/UiO-66@α-Al2O3 Ceramic Membrane Separation and Catalytic Spraying Industry VOCs Waste Gas [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220025. |
| [3] | HAO Honglei, MENG Fanyu, LI Ruoyu, LI Yingqiu, JIA Mingjun, ZHANG Wenxiang, YUAN Xiaoling. Biomass Derived Nitrogen Doped Porous Carbon Materials as Adsorbents for Removal of Methylene Blue in Water [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220055. |
| [4] | WONG Honho, LU Qiuyang, SUN Mingzi, HUANG Bolong. Rational Design of Graphdiyne-based Atomic Electrocatalysts: DFT and Self-validated Machine Learning [J]. Chem. J. Chinese Universities, 2022, 43(5): 20220042. |
| [5] | WANG Hongning, HUANG Li, QING Jiang, MA Tengzhou, JIANG Wei, HUANG Weiqiu, CHEN Ruoyu. Activation of Biochar from Cattail and the VOCs Adsorption Application [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210824. |
| [6] | MENG Xianglong, YANG Ge, GUO Hailing, LIU Chenguang, CHAI Yongming, WANG Chunzheng, GUO Yongmei. Synthesis of Nano-zeolite and Its Adsorption Performance for Hydrogen Sulfide [J]. Chem. J. Chinese Universities, 2022, 43(3): 20210687. |
| [7] | CHEN Xiaolu, YUAN Zhenyan, ZHONG Yingchun, REN Hao. Preparation of Triphenylamine Based PAF-106s via Mechanical Ball Milling and C2 Hydrocarbons Adsorption Property [J]. Chem. J. Chinese Universities, 2022, 43(3): 20210771. |
| [8] | TAN Lejian, ZHONG Xuanshu, WANG Jin, LIU Zongjian, ZHANG Aiying, YE Lin, FENG Zengguo. Low Critical Dissolution Temperature Behavior of β⁃Cyclodextrin and Its Application in the Preparation of β⁃Cyclodextrin Sheet Crystal with Ordered Nano⁃channel [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220405. |
| [9] | LIU Yang, LI Wangchang, ZHANG Zhuxia, WANG Fang, YANG Wenjing, GUO Zhen, CUI Peng. Theoretical Exploration of Noncovalent Interactions Between Sc3C2@C80 and [12]Cycloparaphenylene Nanoring [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220457. |
| [10] | ZHENG Meiqi, MAO Fangqi, KONG Xianggui, DUAN Xue. Layered Double Hydroxides as Sorbent for Remediation of Radioactive Wastewater [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220456. |
| [11] | WANG Yuanyue, AN Suosuo, ZHENG Xuming, ZHAO Yanying. Spectroscopic and Theoretical Studies on 5-Mercapto-1,3,4-thiadiazole-2-thione Microsolvation Clusters [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220354. |
| [12] | TIAN Xiaokang, ZHANG Qingsong, YANG Shulin, BAI Jie, CHEN Bingjie, PAN Jie, CHEN Li, WEI Yen. Porous Materials Inspired by Microbial Fermentation: Preparation Method and Application [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220216. |
| [13] | CHENG Yuanyuan, XI Biying. Theoretical Study on the Fragmentation Mechanism of CH3SSCH3 Radical Cation Initiated by OH Radical [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220271. |
| [14] | ZHOU Chengsi, ZHAO Yuanjin, HAN Meichen, YANG Xia, LIU Chenguang, HE Aihua. Regulation of Silanes as External Electron Donors on Propylene/butene Sequential Polymerization [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220290. |
| [15] | ZHANG Chi, SUN Fuxing, ZHU Guangshan. Synthesis, N2 Adsorption and Mixed-matrix Membrane Performance of Bimetal Isostructural CAU-21 [J]. Chem. J. Chinese Universities, 2022, 43(1): 20210578. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||