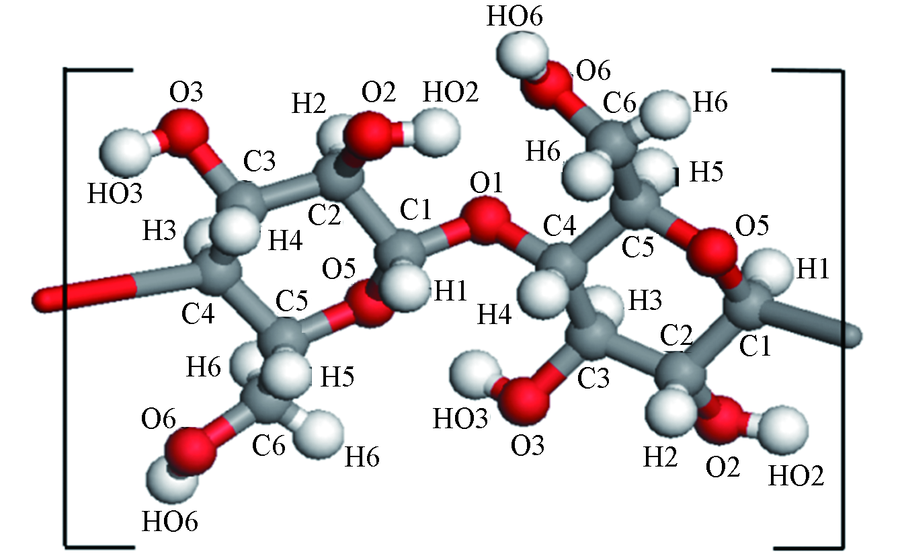
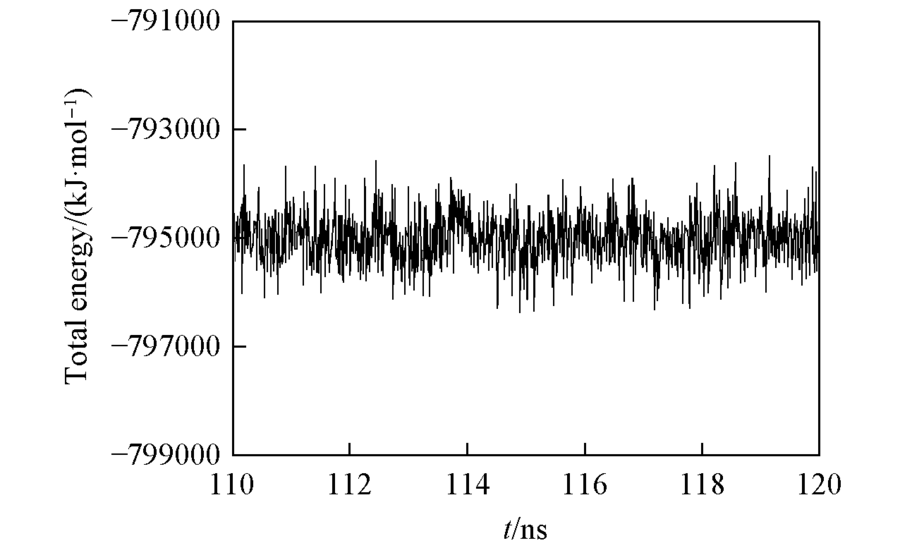
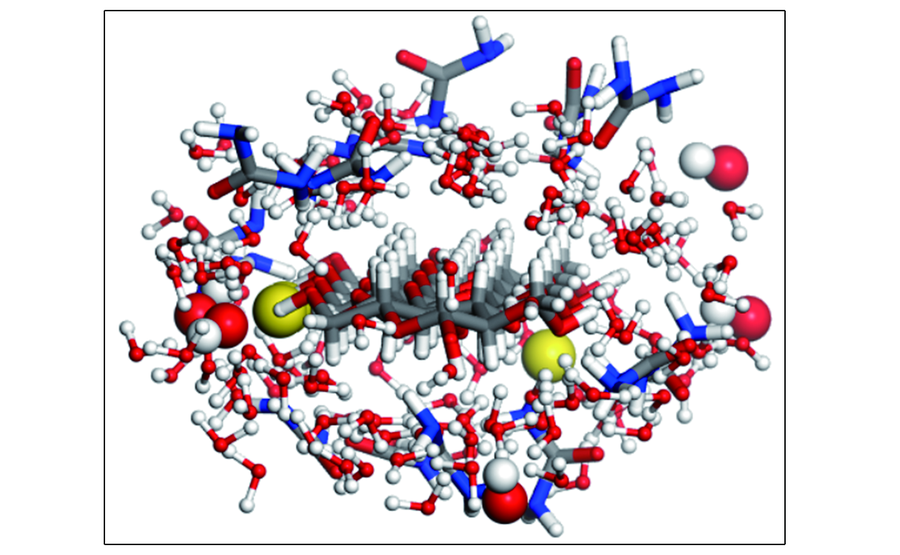
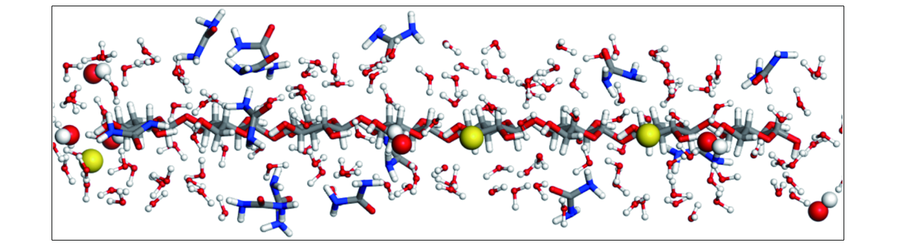
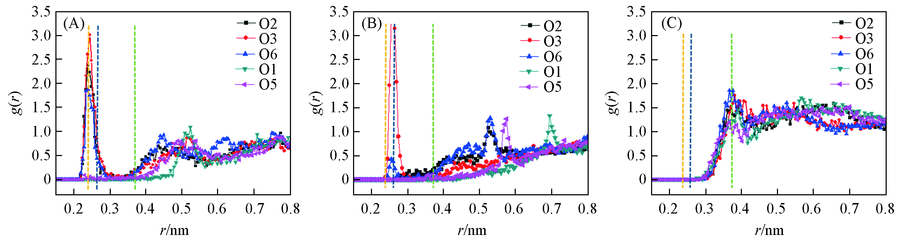
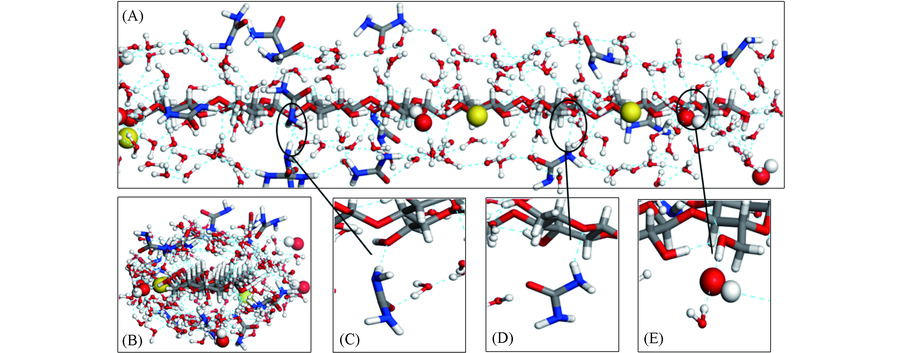
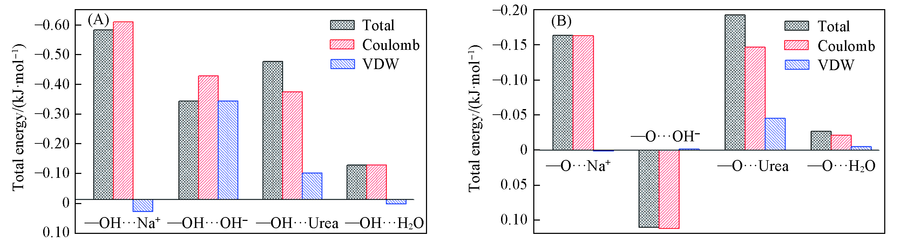
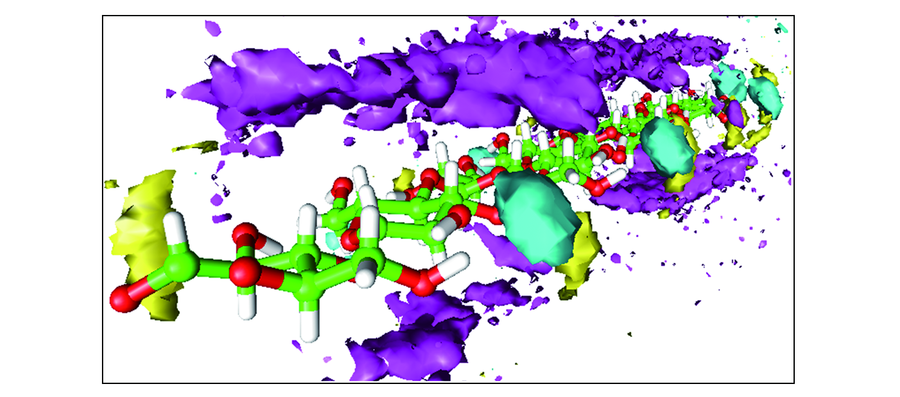
| [1] |
Ghanbari D., Salavati-Niasari M., Beshkar F., Amiri O., J. Mater. Sci-Mater. El., 2015, 26, 8358-8366
|
| [2] |
Vega B., Wondraczek H., Bretschneider L., Nareoja T., Fardim P., Heinze T., Carbohyd. Polym., 2015, 132, 261-273
|
| [3] |
Jiang G. S., Yuan Y., Wang B. C., Mukuze K. S., Huang W. F., Zhang Y. M., Wang H. P., Cellulose,2012, 19, 1075-1083
|
| [4] |
Jimenez de la Parra C., Zambrano J. R., Dolores Bermejo M,, Martin A., Segovia J. J., Cocero M. J., Carbohyd. Polym., 2015, 91, 8-16
|
| [5] |
Luo X. G., Zeng J., Liu S. L., Zhang L. N., Bioresour. Technol., 2015, 194, 403-406
|
| [6] |
Zhou H. Y., Cao P. P., Li J. B., Zhang F. L., Ding P. P., J. Appl. Polym. Sci., 2015, 132, 42152
|
| [7] |
Kolakovic R., Peltonen L., Laukkanen A., Hirvonen J., Laaksonen T., Eur. J. Pharm. Biopharm., 2012, 82, 308-315
|
| [8] |
He M., Zhao Y., Duan J., Wang Z., Chen Y., Zhang L., ACS Appl. Mat. Interfaces, 2014, 6, 1872-1878
|
| [9] |
Huang C. F., Chen J. K., Tsai T. Y., Hsieh Y. A., Lin K. Y. A., Polymer,2015, 72, 195-200
|
| [10] |
Han H., Liang Y., Wang F., Zhang Y., Food Science and Technology,2011, 36, 48-53
|
|
(韩浩, 梁琰, 王凤, 张勇. 食品科技, 2011, 36, 50-53)
|
| [11] |
Liu T., Liu N., Fang G. Z., Food Science, 2009, 30, 376-280
|
|
(刘涛, 刘宁, 方桂珍. 食品科学, 2009, 30, 276-280)
|
| [12] |
Fink H. P., Weigel P., Purz H. J., Ganster J., Prog. Polym. Sci., 2001, 26, 1473-1524
|
| [13] |
Liu R. G., Shen Y. Y., Shao H. L., Wu C. X., Hu X. C., Cellulose,2001, 8, 13-21
|
| [14] |
Morgenstern B., Kammer H. W., Berger W., Skrabal P., Acta Poly., 1992, 43, 356-357
|
| [15] |
Zhang C., Liu R. G., Xiang J. F., Kang H. L., Liu Z. J., Huang Y., J. Phys. Chem. B, 2014, 118, 9507-9514
|
| [16] |
Wang H., Gurau G., Rogers R. D., Chem. Soc. Rev., 2012, 41, 1519-1537
|
| [17] |
Aaltonen O., Jauhiainen O., Carbohyd. Polym., 2009, 75, 125-129
|
| [18] |
Cai J., Zhang L., Zhou J., Li H., Chen H., Jin H., Macromol. Rapid Comm., 2004, 25, 1558-1562
|
| [19] |
Cai J., Zhang L., Biomacromolecules,2006, 7, 183-189
|
| [20] |
Cai J., Zhang L., Zhou J., Qi H., Chen H., Kondo T., Chen X., Chu B., Adv. Mater., 2007, 19, 821-825
|
| [21] |
Cai J., Zhang L., Chang C., Cheng G., Chen X., Chu B., Chem. Phys. Chem., 2007, 8, 1572-1579
|
| [22] |
Lue A., Zhang L., Ruan D., Macromol. Chem. Phys., 2007, 208, 2359-2366
|
| [23] |
Cai J., Zhang L., Macromol. Biosci., 2005, 5, 539-548
|
| [24] |
Cai J., Zhang L., Liu S., Liu Y., Xu X., Chen X., Chu B., Guo X., Xu J., Cheng H., Macromolecules,2008, 41, 9345-9351
|
| [25] |
Qin X., Lu A., Cai J., Zhang L., Carbohyd. Polym., 2013, 92, 1315-1320
|
| [26] |
Jiang Z., Fang Y., Xiang J., Ma Y., Lu A., Kang H., Huang Y., Guo H., Liu R., Zhang L., J. Phys. Chem. B, 2014, 118, 10250-10257
|
| [27] |
Cai L., Liu Y., Liang H., Polymer,2012, 53, 1124-1130
|
| [28] |
Wernersson E., Stenqvist B., Lund M., Cellulose,2015, 22, 991-1001
|
| [29] |
Miyamoto H., Ueda K., Yamane C., Nord. Pulp Pap. Res. J., 2015, 30, 67-77
|
| [30] |
Guvench O., Hatcher E., Venable R. M., Pastor R. W., MacKerell A. D. Jr., J. Chem. Theory Comput., 2009, 5, 2353-2370
|
| [31] |
Weerashinghe S., Smith P. E., J. Phys. Chem. B, 2003, 7, 3891-3898
|
| [32] |
Hess B., Kutzner C., van der Spoel D., Lindahl E., J. Chem. Theory Comput., 2008, 4,435-447
|
| [33] |
Durell S. R., Brooks B. R., Ben-Naim A., J. Chem. Phys., 1994, 98, 2198-2202
|
| [34] |
Gomes T. C., Skaf M. S., J. Comput. Chem., 2012, 33, 1338-1346
|
 ), 苑世领1(
), 苑世领1(