

Chem. J. Chinese Universities ›› 2014, Vol. 35 ›› Issue (6): 1267.doi: 10.7503/cjcu20140046
• Physical Chemistry • Previous Articles Next Articles
GONG Jian1, CAO Hongyu2, LI Shenmin1,2, TANG Qian2, YANG Yanjie2, ZHENG Xuefang1,2,*( )
)
Received:2014-01-15
Online:2014-06-10
Published:2014-04-21
Contact:
ZHENG Xuefang
E-mail:dlxfzheng@126.com
Supported by:CLC Number:
TrendMD:
GONG Jian, CAO Hongyu, LI Shenmin, TANG Qian, YANG Yanjie, ZHENG Xuefang. Theoritical Studies on the Structure and Absorption Spectra of Neo-Confused Metal Porphyrin†[J]. Chem. J. Chinese Universities, 2014, 35(6): 1267.
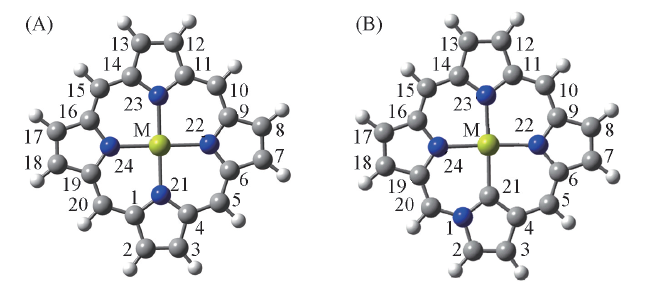
Fig.1 Molecular structures of M-FBP(A) and M-NECP(B) (M: Mg, Ni, Cu, Zn)
| Species | Mg-FBP | Ni-FBP | Cu-FBP | Zn-FBP |
|---|---|---|---|---|
| M—N21/nm | 0.2066(0.2052[ | 0.1975(0.1972[ | 0.2023(0.2007[ | 0.2056(0.2058[ |
| N21—C4/nm | 0.1374(0.1366[ | 0.1378(0.1380[ | 0.1375(0.1376[ | 0.1373(0.1372[ |
| C4—C3/nm | 0.1448(0.1443[ | 0.1441(0.1438[ | 0.1445(0.1443[ | 0.1447(0.1446[ |
| C3—C2/nm | 0.1367(0.1354[ | 0.1360(0.1360[ | 0.1363(0.1361[ | 0.1366(0.1366[ |
| C4—C5/nm | 0.1402(0.1386[ | 0.1383(0.1380[ | 0.1391(0.1388[ | 0.1398(0.1398[ |
| N24—M—N21/(°) | 90.00 | 90.00(90.00[ | 90.00 | 90.00(90.00[ |
| N21—C1—C2/(°) | 109.50(108.9[ | 111.02(110.93[ | 110.13 | 109.58(109.52[ |
| C2—C3—C4/(°) | 106.93(107.2[ | 106.61(106.68[ | 106.79 | 106.91(106.92[ |
| C1—N21—C4/(°) | 107.14(107.8[ | 104.74(104.79[ | 106.17 | 107.02(107.13[ |
| C20—C1—N21/(°) | 124.95(125.6[ | 125.45(125.53[ | 125.31 | 125.05(125.07[ |
| C4—C5—C6/(°) | 127.24(126.6[ | 123.85(123.72[ | 125.55 | 126.92(126.99[ |
Table 1 Geometric parameters of M-FBP(M: Mg, Ni, Cu and Zn) at B3LYP/6-31+G(d) level
| Species | Mg-FBP | Ni-FBP | Cu-FBP | Zn-FBP |
|---|---|---|---|---|
| M—N21/nm | 0.2066(0.2052[ | 0.1975(0.1972[ | 0.2023(0.2007[ | 0.2056(0.2058[ |
| N21—C4/nm | 0.1374(0.1366[ | 0.1378(0.1380[ | 0.1375(0.1376[ | 0.1373(0.1372[ |
| C4—C3/nm | 0.1448(0.1443[ | 0.1441(0.1438[ | 0.1445(0.1443[ | 0.1447(0.1446[ |
| C3—C2/nm | 0.1367(0.1354[ | 0.1360(0.1360[ | 0.1363(0.1361[ | 0.1366(0.1366[ |
| C4—C5/nm | 0.1402(0.1386[ | 0.1383(0.1380[ | 0.1391(0.1388[ | 0.1398(0.1398[ |
| N24—M—N21/(°) | 90.00 | 90.00(90.00[ | 90.00 | 90.00(90.00[ |
| N21—C1—C2/(°) | 109.50(108.9[ | 111.02(110.93[ | 110.13 | 109.58(109.52[ |
| C2—C3—C4/(°) | 106.93(107.2[ | 106.61(106.68[ | 106.79 | 106.91(106.92[ |
| C1—N21—C4/(°) | 107.14(107.8[ | 104.74(104.79[ | 106.17 | 107.02(107.13[ |
| C20—C1—N21/(°) | 124.95(125.6[ | 125.45(125.53[ | 125.31 | 125.05(125.07[ |
| C4—C5—C6/(°) | 127.24(126.6[ | 123.85(123.72[ | 125.55 | 126.92(126.99[ |
| Species | Mg-NECP | Ni-NECP | Cu-NECP | Zn-NECP |
|---|---|---|---|---|
| M—C21/nm | 0.2085 | 0.1923(0.1907[ | 0.1975 | 0.1990 |
| M—N22/nm | 0.2074 | 0.1984(0.1951[ | 0.2050 | 0.2106 |
| M—N23/nm | 0.2036 | 0.1991(0.1970[ | 0.2014 | 0.2017 |
| M—N24/nm | 0.2070 | 0.1954(0.1929[ | 0.2031 | 0.2105 |
| C21—N1/nm | 0.1373 | 0.1385(0.1399[ | 0.1379 | 0.1379 |
| C21—C4/nm | 0.1411 | 0.1419(0.1402[ | 0.1413 | 0.1412 |
| N23—C1/nm | 0.1376 | 0.1377(0.1367[ | 0.1375 | 0.1379 |
| N23—C14/nm | 0.1371 | 0.1375(0.1384[ | 0.1373 | 0.1378 |
| N24—M—C21/(°) | 88.67 | 90.15 | 89.97 | 90.13 |
| C21—N1—C2/(°) | 110.95 | 112.32 | 111.36 | 110.69 |
| C2—C3—C4/(°) | 107.05 | 106.70 | 106.97 | 107.18 |
| N1—C21—C4/(°) | 105.85 | 103.15 | 104.81 | 105.79 |
| C20—N1—C21/(°) | 125.22 | 126.86 | 126.73 | 126.68 |
| C4—C5—C6/(°) | 127.29 | 122.73 | 124.93 | 126.65 |
Table 2 Geometric parameters of M-NECP(M: Mg, Ni, Cu, Zn) at B3LYP/6-31+G(d) level
| Species | Mg-NECP | Ni-NECP | Cu-NECP | Zn-NECP |
|---|---|---|---|---|
| M—C21/nm | 0.2085 | 0.1923(0.1907[ | 0.1975 | 0.1990 |
| M—N22/nm | 0.2074 | 0.1984(0.1951[ | 0.2050 | 0.2106 |
| M—N23/nm | 0.2036 | 0.1991(0.1970[ | 0.2014 | 0.2017 |
| M—N24/nm | 0.2070 | 0.1954(0.1929[ | 0.2031 | 0.2105 |
| C21—N1/nm | 0.1373 | 0.1385(0.1399[ | 0.1379 | 0.1379 |
| C21—C4/nm | 0.1411 | 0.1419(0.1402[ | 0.1413 | 0.1412 |
| N23—C1/nm | 0.1376 | 0.1377(0.1367[ | 0.1375 | 0.1379 |
| N23—C14/nm | 0.1371 | 0.1375(0.1384[ | 0.1373 | 0.1378 |
| N24—M—C21/(°) | 88.67 | 90.15 | 89.97 | 90.13 |
| C21—N1—C2/(°) | 110.95 | 112.32 | 111.36 | 110.69 |
| C2—C3—C4/(°) | 107.05 | 106.70 | 106.97 | 107.18 |
| N1—C21—C4/(°) | 105.85 | 103.15 | 104.81 | 105.79 |
| C20—N1—C21/(°) | 125.22 | 126.86 | 126.73 | 126.68 |
| C4—C5—C6/(°) | 127.29 | 122.73 | 124.93 | 126.65 |
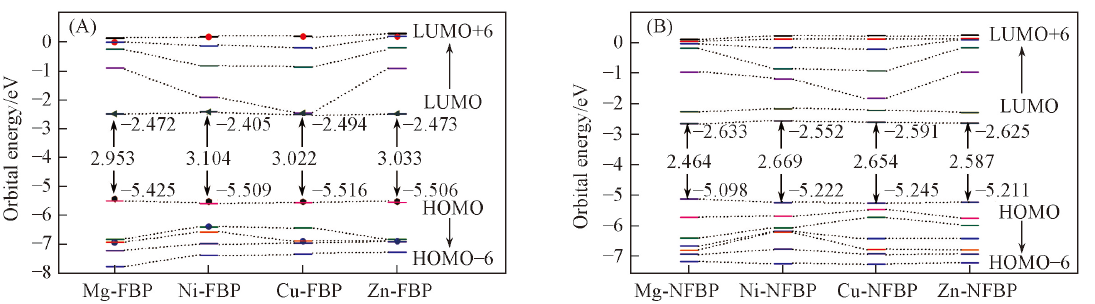
Fig.2 Orbital energy levels and the ΔE(HOMO-LUMO)/eV of M-FBP(A) and M-NECP(B)
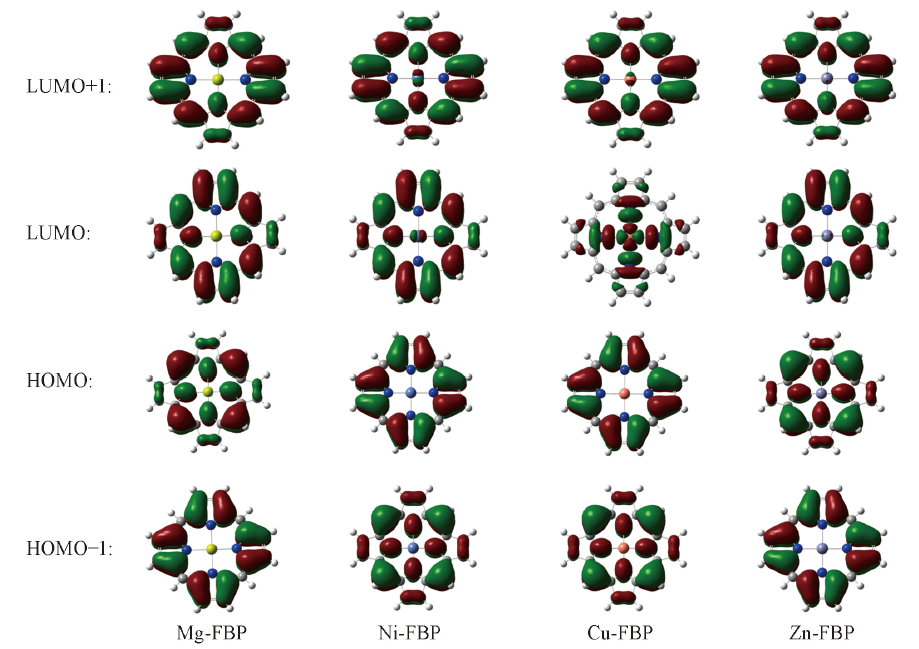
Fig.3 Frontier molecule orbitals distributions of M-FBP(M: Mg, Ni, Cu, Zn)
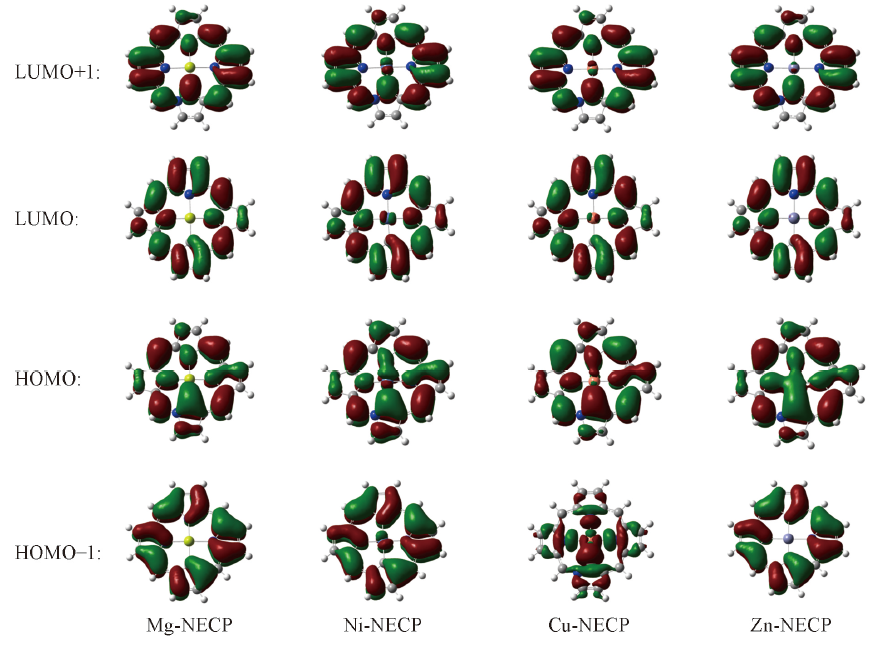
Fig.4 Frontier molecule orbitals distributions of M-NECP(M: Mg, Ni, Cu, Zn)
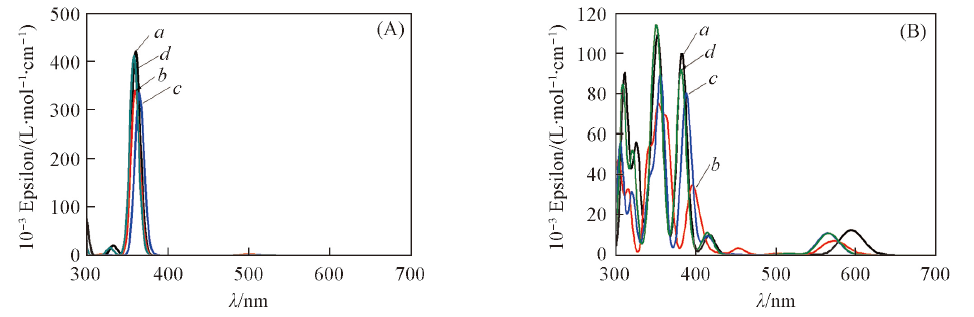
Fig.5 Simulated absorption spectrum of M-FBP(A) and M-NECP(B) a. Mg; b. Ni; c. Cu; d. Zn.
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Mg-FBP | Eu | 2.348 | 528 | 0.0007 | H-1→L(47), H→L+1(52) |
| Eu | 2.348 | 528 | 0.0007 | H-1→L+1(47), H→L(52) | |
| Eu | 3.451 | 359 | 0.9685 | H-1→L(49), H→L+1(43) | |
| Eu | 3.451 | 359 | 0.9685 | H-1→L+1(49), H→L(43) | |
| Eu | 3.738 | 332 | 0.0474 | H-2→L(87) | |
| Eu | 3.738 | 332 | 0.0474 | H-2→L+1(87) | |
| Mg-NECP | A | 2.088 | 594 | 0.0578 | H-1→L+1(10), H→L(84) |
| A | 2.342 | 529 | 0.0026 | H-1→L(39), H→L+1(56) | |
| A | 2.980 | 416 | 0.0465 | H-2→L(51), H-1→L+1(41) | |
| A | 3.249 | 382 | 0.4637 | H-2→L+1(17), H-1→L(34), H→L+1(25) | |
| A | 3.504 | 354 | 0.3420 | H-4→L(17), H-2→L(33), H→L+2(17) | |
| A | 3.814 | 325 | 0.2335 | H-6→L(17), H-5→L(23), H→L+2(35) |
Table 3 Excitation energy levels, oscillators strength(f) and transition configurations for Mg-FBP and Mg-NECP molecules in the gas state
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Mg-FBP | Eu | 2.348 | 528 | 0.0007 | H-1→L(47), H→L+1(52) |
| Eu | 2.348 | 528 | 0.0007 | H-1→L+1(47), H→L(52) | |
| Eu | 3.451 | 359 | 0.9685 | H-1→L(49), H→L+1(43) | |
| Eu | 3.451 | 359 | 0.9685 | H-1→L+1(49), H→L(43) | |
| Eu | 3.738 | 332 | 0.0474 | H-2→L(87) | |
| Eu | 3.738 | 332 | 0.0474 | H-2→L+1(87) | |
| Mg-NECP | A | 2.088 | 594 | 0.0578 | H-1→L+1(10), H→L(84) |
| A | 2.342 | 529 | 0.0026 | H-1→L(39), H→L+1(56) | |
| A | 2.980 | 416 | 0.0465 | H-2→L(51), H-1→L+1(41) | |
| A | 3.249 | 382 | 0.4637 | H-2→L+1(17), H-1→L(34), H→L+1(25) | |
| A | 3.504 | 354 | 0.3420 | H-4→L(17), H-2→L(33), H→L+2(17) | |
| A | 3.814 | 325 | 0.2335 | H-6→L(17), H-5→L(23), H→L+2(35) |
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Ni-FBP | Eu | 2.478 | 500 | 0.0059 | H-1→L(46), H→L+1(54) |
| Eu | 2.478 | 500 | 0.0059 | H-1→L+1(46), H→L(54) | |
| Eu | 3.456 | 359 | 0.7851 | H-1→L+1(51), H→L(45) | |
| Eu | 3.456 | 359 | 0.7852 | H-1→L(51), H→L+1(45) | |
| Eu | 3.919 | 316 | 0.006 | H-5→L(96) | |
| Eu | 3.919 | 316 | 0.006 | H-5→L+1(96) | |
| Ni-NECP | A | 2.133 | 581 | 0.0161 | H-3→L+2(48), H→L(40) |
| A | 2.188 | 567 | 0.0222 | H-3→L+2(45), H→L(42) | |
| A | 3.150 | 394 | 0.145 | H-4→L+1(34), H-1→L(10), H-1→L+1(24) | |
| A | 3.529 | 351 | 0.3046 | H-4→L+1(34), H-2→L+1(25), H-1→L(13) | |
| A | 3.655 | 339 | 0.2265 | H-5→L(56), H-1→L+1(13) |
Table 4 Excitation energy levels, oscillators strength(f) and transition configurations for Ni-FBP and Ni-NECP molecules in the gas state
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Ni-FBP | Eu | 2.478 | 500 | 0.0059 | H-1→L(46), H→L+1(54) |
| Eu | 2.478 | 500 | 0.0059 | H-1→L+1(46), H→L(54) | |
| Eu | 3.456 | 359 | 0.7851 | H-1→L+1(51), H→L(45) | |
| Eu | 3.456 | 359 | 0.7852 | H-1→L(51), H→L+1(45) | |
| Eu | 3.919 | 316 | 0.006 | H-5→L(96) | |
| Eu | 3.919 | 316 | 0.006 | H-5→L+1(96) | |
| Ni-NECP | A | 2.133 | 581 | 0.0161 | H-3→L+2(48), H→L(40) |
| A | 2.188 | 567 | 0.0222 | H-3→L+2(45), H→L(42) | |
| A | 3.150 | 394 | 0.145 | H-4→L+1(34), H-1→L(10), H-1→L+1(24) | |
| A | 3.529 | 351 | 0.3046 | H-4→L+1(34), H-2→L+1(25), H-1→L(13) | |
| A | 3.655 | 339 | 0.2265 | H-5→L(56), H-1→L+1(13) |
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Cu-FBP | Eu | 2.424 | 511 | 0.0030 | H-1(α)→L+1(α)(25), H(α)→L(α)(23), H-1(β)→L+1(β)(28), |
| H(β)→L+2(β)(23) | |||||
| Eu | 2.424 | 511 | 0.0030 | H-1(α)→L(α)(25), H(α)→L+1(α)(23), H-1(β)→L+2(β)(28), | |
| H(β)→L+1(β)(23) | |||||
| Eu | 3.285 | 377 | 0.0055 | H-5(α)→L+1(α)(35), H-4(β)→L+2(β)(40) | |
| Eu | 3.285 | 377 | 0.0055 | H-5(α)→L(α)(35), H-4(β)→L+1(β)(40) | |
| Eu | 3.409 | 364 | 0.7721 | H-1(α)→L(α)(24), H(α)→L+1(α)(22), H-1(β)→L+2(β)(24), | |
| H(β)→L+1(β)(23) | |||||
| Eu | 3.409 | 364 | 0.7721 | H-1(α)→L+1(α)(24), H(α)→L(α)(22), H-1(β)→L+1(β)(24), | |
| H(β)→L+2(β)(23) | |||||
| Cu-NECP | A | 2.196 | 565 | 0.0506 | H(α)→L(α)(41), H(β)→L(β)(41) |
| A | 2.353 | 527 | 0.0001 | H-1(α)→L+1(α)(27), H(β)→L+2(β)(68) | |
| A | 2.993 | 414 | 0.0392 | H-3(α)→L(α)(27), H-2(α)→L+1(α)(14), H-2(β)→L(β)(19), | |
| H-1(β)→L+1(β)(15) | |||||
| A | 3.204 | 387 | 0.3423 | H(α)→L+1(α)(12), H-1(β)→L(β)(14) | |
| A | 3.493 | 355 | 0.3301 | H-4(α)→L(α)(12), H-3(α)→L(α)(18), H-2(β)→L(β)(18) | |
| A | 3.887 | 319 | 0.1371 | H(α)→L+2(α)(38), H(β)→L+3(β)(33) |
Table 5 Excitation energy levels, oscillators strength(f) and transition configurations for Cu-FBP and Cu-NECP molecules in the gas state
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Cu-FBP | Eu | 2.424 | 511 | 0.0030 | H-1(α)→L+1(α)(25), H(α)→L(α)(23), H-1(β)→L+1(β)(28), |
| H(β)→L+2(β)(23) | |||||
| Eu | 2.424 | 511 | 0.0030 | H-1(α)→L(α)(25), H(α)→L+1(α)(23), H-1(β)→L+2(β)(28), | |
| H(β)→L+1(β)(23) | |||||
| Eu | 3.285 | 377 | 0.0055 | H-5(α)→L+1(α)(35), H-4(β)→L+2(β)(40) | |
| Eu | 3.285 | 377 | 0.0055 | H-5(α)→L(α)(35), H-4(β)→L+1(β)(40) | |
| Eu | 3.409 | 364 | 0.7721 | H-1(α)→L(α)(24), H(α)→L+1(α)(22), H-1(β)→L+2(β)(24), | |
| H(β)→L+1(β)(23) | |||||
| Eu | 3.409 | 364 | 0.7721 | H-1(α)→L+1(α)(24), H(α)→L(α)(22), H-1(β)→L+1(β)(24), | |
| H(β)→L+2(β)(23) | |||||
| Cu-NECP | A | 2.196 | 565 | 0.0506 | H(α)→L(α)(41), H(β)→L(β)(41) |
| A | 2.353 | 527 | 0.0001 | H-1(α)→L+1(α)(27), H(β)→L+2(β)(68) | |
| A | 2.993 | 414 | 0.0392 | H-3(α)→L(α)(27), H-2(α)→L+1(α)(14), H-2(β)→L(β)(19), | |
| H-1(β)→L+1(β)(15) | |||||
| A | 3.204 | 387 | 0.3423 | H(α)→L+1(α)(12), H-1(β)→L(β)(14) | |
| A | 3.493 | 355 | 0.3301 | H-4(α)→L(α)(12), H-3(α)→L(α)(18), H-2(β)→L(β)(18) | |
| A | 3.887 | 319 | 0.1371 | H(α)→L+2(α)(38), H(β)→L+3(β)(33) |
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Zn-FBP | Eu | 2.397 | 517 | 0.0021 | H-1→L(49), H→L+1(49) |
| Eu | 2.397 | 517 | 0.0021 | H-1→L+1(49), H→L(49) | |
| Eu | 3.472 | 357 | 0.9494 | H-1→LO(13), H-1→L+1(36), H→L(35), H→L+1(12) | |
| Eu | 3.472 | 357 | 0.9494 | H-1→L(36), H-1→L+1(13), H→L+1(35) | |
| Eu | 3.786 | 327 | 0.0376 | H-3→L+1(90) | |
| Eu | 3.786 | 327 | 0.0376 | H-3→L(90) | |
| Zn-NECP | A | 2.193 | 565 | 0.0502 | H-1→L+1(11), H→L(79) |
| A | 2.406 | 515 | 0.0036 | H-1→L(40), H→L+1(52) | |
| A | 2.998 | 414 | 0.0518 | H-3→LUMO(52), H-1→L+1(37) | |
| A | 3.258 | 381 | 0.4238 | H-3→L+1(17), H-1→L(30), H-1→L+1(17), H→L+1(25) | |
| A | 3.562 | 348 | 0.2447 | H-5→L(21), H-4→L(38) | |
| A | 3.875 | 320 | 0.2171 | H-4→L+1(15), H→L+2(46) |
Table 6 Excitation energy levels, oscillators strength(f) and transition configurations for Zn-FBP and Zn-NECP molecules in the gas state
| Molecular | State | Excitation energy/eV | λ/nm | f | Main configuration(%) |
|---|---|---|---|---|---|
| Zn-FBP | Eu | 2.397 | 517 | 0.0021 | H-1→L(49), H→L+1(49) |
| Eu | 2.397 | 517 | 0.0021 | H-1→L+1(49), H→L(49) | |
| Eu | 3.472 | 357 | 0.9494 | H-1→LO(13), H-1→L+1(36), H→L(35), H→L+1(12) | |
| Eu | 3.472 | 357 | 0.9494 | H-1→L(36), H-1→L+1(13), H→L+1(35) | |
| Eu | 3.786 | 327 | 0.0376 | H-3→L+1(90) | |
| Eu | 3.786 | 327 | 0.0376 | H-3→L(90) | |
| Zn-NECP | A | 2.193 | 565 | 0.0502 | H-1→L+1(11), H→L(79) |
| A | 2.406 | 515 | 0.0036 | H-1→L(40), H→L+1(52) | |
| A | 2.998 | 414 | 0.0518 | H-3→LUMO(52), H-1→L+1(37) | |
| A | 3.258 | 381 | 0.4238 | H-3→L+1(17), H-1→L(30), H-1→L+1(17), H→L+1(25) | |
| A | 3.562 | 348 | 0.2447 | H-5→L(21), H-4→L(38) | |
| A | 3.875 | 320 | 0.2171 | H-4→L+1(15), H→L+2(46) |
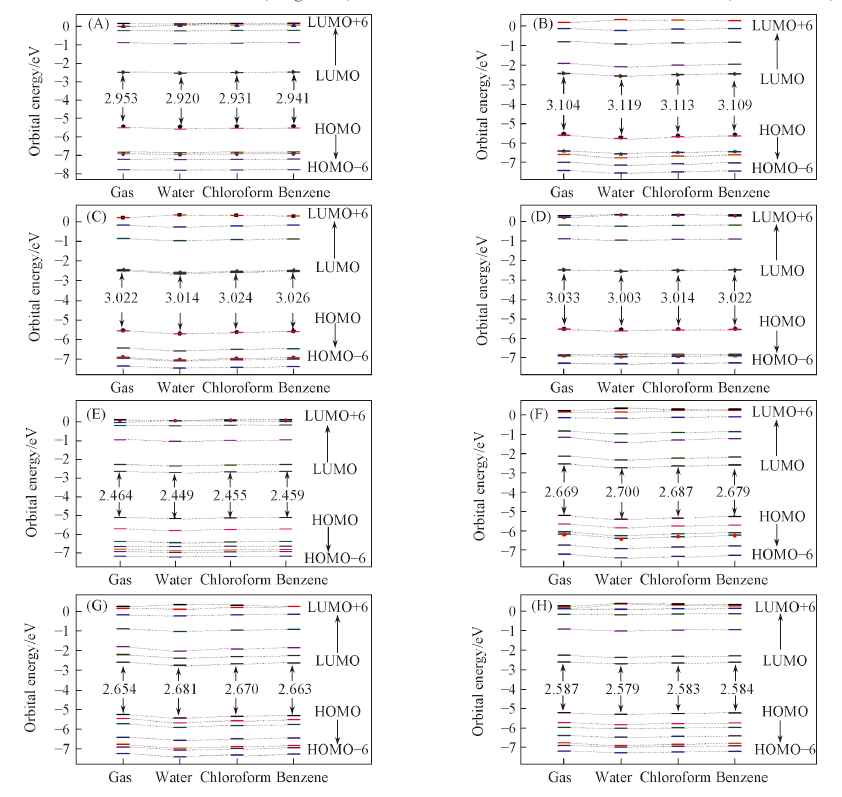
Fig.6 Orbital energy levels and the ΔE(HOMO-LUMO)/eV of Mg-FBP(A), Ni-FBP(B), Cu-FBP(C), Zn-FBP(D), Mg-NECP(E), Ni-NECP(F), Cu-NECP(G) and Zn-NECP(H) in gas and solvents(water, chloroform and benzene)
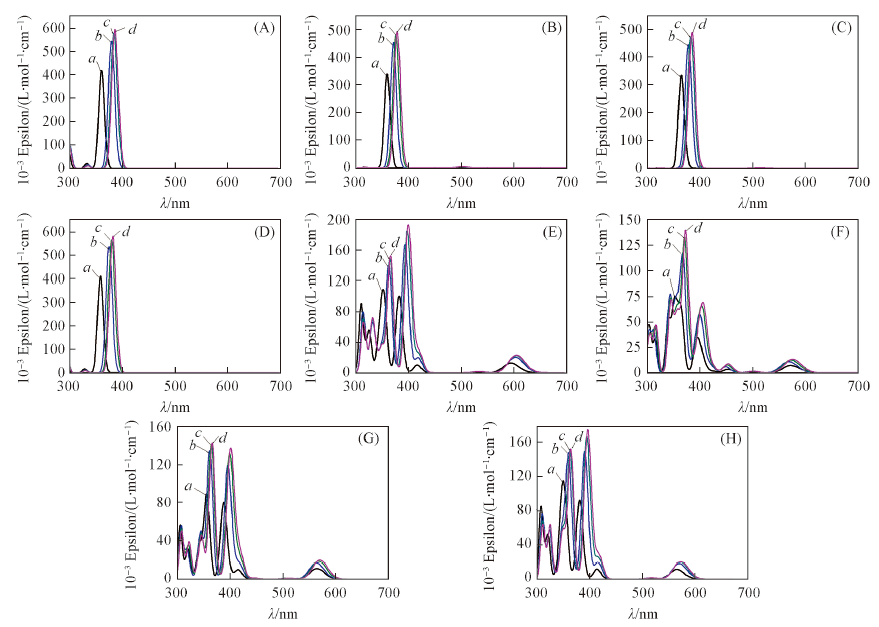
Fig.7 Simulated absorption spectrum of Mg-FBP(A), Ni-FBP(B), Cu-FBP(C), Zn-FBP(D), Mg-NECP(E), Ni-NECP(F), Cu-NECP(G)and Zn-NECP(H) in gas(a), water(b), chloroform(c), benzene(d)
| [1] | Smith K.M., Porphyrins and Metalloporphyrins, Elsevier, Amsterdam, 1975 |
| [2] | Battersby A. R., Fookes C. J., Matcham G. W., McDonald E., Nature, 1980, 285(5759), 17—21 |
| [3] | Kräutler B., Chimia, 1987, 41(9), 277—292 |
| [4] | Kay A., Graetzel M., J. Phys. Chem., 1993, 97(23), 6272—6277 |
| [5] | Kay A., Humphry-Baker R., Graetzel M., J. Phys. Chem., 1994, 98(3), 952—959 |
| [6] | Beljonne D., O'Keefe G. E., Hamer P. J., Friend R. H., Anderson H. L., Brédas J. L., J. Chem. Phys., 1997, 106(23), 9439—9460 |
| [7] | Henari F. Z., Blau W. J., Milgrom L. R., Yahioglu G., Phillips D., Lacey J. A., Chem. Phys. Lett., 1997, 267(3/4), 229—233 |
| [8] | Wang Z. Q., Day P. N., Pachter R ., J. Chem. Phys., 1998, 108(6), 2504—2510 |
| [9] | Merchán M., Ortí E., Roos B. O., Chem. Phys. Lett., 1994, 226(1/2), 27—36 |
| [10] | Nakatsuji H., Hasegawa J. Y., Hada M., J. Chem. Phys., 1996, 104(6), 2321—2329 |
| [11] | Kitao O., Ushiyama H., Miura N., J. Chem. Phys., 1999, 110(6), 2936—2946 |
| [12] | van Gisbergen S. J. A., Rosa A., Ricciardi G., Baerends E. J., J. Chem. Phys., 1999, 111(6), 2499—2506 |
| [13] | Sundholm D., Phys. Chem. Chem. Phys., 2000, 2(10), 2275—2281 |
| [14] | Šeda J., Burda J. V., Brázdová V., Int. J. Mol. Sci., 2004, 5(4), 196—213 |
| [15] | Šeda J., Burda J. V., Leszczynski J., J. Comput. Chem., 2005, 26(3), 294—303 |
| [16] | Zhang Y. H., Ruan W. J., Li Z. Y., Wu Y., Zheng J. Y., Chem. Phys., 2005, 315(1/2), 201—213 |
| [17] | Gouterman M., J. Mol. Spectrosc., 1961, 6(0), 138—163 |
| [18] | Petke J. D., Maggiora G. M., Shipman L. L., Christoffersen R. E., J. Mol. Spectrosc., 1978, 71(1—3), 64—84 |
| [19] | Balanay M. P., Kim D. H., J. Mol. Struct.(Theochem.), 2009, 910(1—3), 20—26 |
| [20] | Lash T. D., Lammer A. D., Ferrence G. M., Angew. Chem. Int. Ed., 2011, 50(41), 9718—9721 |
| [21] | Fujino K., Hirata Y., Kawabe Y., Morimoto T., Srinivasan A., Toganoh M., Miseki Y., Kudo A., Furuta H., Angew. Chem. Int. Ed., 2011, 50(30), 6855—6859 |
| [22] | Bauernschmitt R., Ahlrichs R., Chem. Phys. Lett., 1996, 256(4/5), 454—464 |
| [23] | Stratmann R. E., Scuseria G. E., Frisch M. J., J. Chem. Phys., 1998, 109(19), 8218—8224 |
| [24] | Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A., Gaussian 03, Revision B.04, Wallingford CT, Gaussian Inc., 2004 |
| [25] | Becke A. D., Phys. Rev. A, 1988, 38(6), 3098—3100 |
| [26] | Lee C., Yang W., Parr R. G., Phys. Rev. B, 1988, 37(2), 785—789 |
| [27] | O’Boyle N. M., Tenderholt A. L., Langner K. M., J. Comput. Chem., 2008, 29(5), 839—845 |
| [28] | Wei L., She Y. B., Yu Y. M., Yao X. Q., Zhang S. J., J. Mol .Model., 2012, 18(6), 2483—2491 |
| [29] | Liu X., Yeow E. K. L., Velate S., Steer R. P., Phys. Chem. Chem. Phys., 2006, 8(11), 1298—1309 |
| [30] | Hashimoto T., Choe Y. K., Nakano H., Hirao K., J. Phys. Chem. A, 1999, 103(12), 1894—1904 |
| [31] | Ghosh A., Vangberg T., Inorg. Chem., 1998, 37(24), 6276—6280 |
| [32] | Hirao H., Shaik S., Kozlowski P. M., J. Phys. Chem. A, 2006, 110(18), 6091—6099 |
| [1] | HE Hongrui, XIA Wensheng, ZHANG Qinghong, WAN Huilin. Density-functional Theoretical Study on the Interaction of Indium Oxyhydroxide Clusters with Carbon Dioxide and Methane [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220196. |
| [2] | WANG Yuanyue, AN Suosuo, ZHENG Xuming, ZHAO Yanying. Spectroscopic and Theoretical Studies on 5-Mercapto-1,3,4-thiadiazole-2-thione Microsolvation Clusters [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220354. |
| [3] | YING Fuming, JI Chenru, SU Peifeng, WU Wei. λ-DFCAS: A Hybrid Density Functional Complete Active Space Self Consistent Field Method [J]. Chem. J. Chinese Universities, 2021, 42(7): 2218. |
| [4] | CAO Hongyu,MA Zihui,ZHANG Wenqiong,TANG Qian,LI Ruyu,ZHENG Xuefang. Structures and Electronic Absorption Spectra of N/Neo-Confused, Doubly N-Confused and Neo-Confused N-Confused Porphyrin Isomers † [J]. Chem. J. Chinese Universities, 2020, 41(2): 341. |
| [5] | WEI Xin, DENG Yaoliang, ZHENG Xuming, ZHAO Yanying. Ground Structure and Excited State Proton Transfer Reaction of 2-Aminobenzothiazole [J]. Chem. J. Chinese Universities, 2019, 40(8): 1679. |
| [6] | LIU Qiuna,XU Wenwen,LIU Maozhu,WANG Huigang,ZHENG Xuming. Study on Raman Spectroscopy Non-coincidence Effect of Propionic Anhydride C=O Vibration Mode† [J]. Chem. J. Chinese Universities, 2019, 40(5): 932. |
| [7] | ZHI Shasha,BAN Ying,XU Zhiguang,XU Xuan. Electron Transport of Metal String Complexes of [MM'M″(dpa)4(Cl)2](M=Co, Ni; M',M″=Co, Rh)† [J]. Chem. J. Chinese Universities, 2019, 40(5): 980. |
| [8] |
ZHOU Xiaofeng,ZHOU Yanbing,TANG Chunmei.
Hydrogen Storage Capacity of the Alkaline Earth Metal Mg Exohedral Doped Boron Cage B40M |
| [9] | ZHOU Hegen,JIN Hua,GUO Huirui,LIN Jing,ZHANG Yongfan. Electronic Structures and Optical Properties of Cu-based Semiconductors with Chalcopyrite-type Structure† [J]. Chem. J. Chinese Universities, 2019, 40(3): 518. |
| [10] | YANG Yejin,YOU Jinglin,WANG Jian,WANG Min,HE Yingxia,WU Zhidong. In-situ High Temperature Raman Spectroscopic and Decomposition Thermodynamic Study of the Structure of Potassium Hydrogen Sulfate and Its Melt† [J]. Chem. J. Chinese Universities, 2018, 39(10): 2272. |
| [11] | WANG Junkai, HAN Lei, HUANG Liang, ZHANG Haijun, LI Junyi, LI Saisai. Density Functional Theory Calculation and Experimental Study of Catalytic Synthesis SiC Nano Powders† [J]. Chem. J. Chinese Universities, 2017, 38(9): 1602. |
| [12] | ZHANG Hui, ZHANG Hongmei, WANG Lianjun, SHEN Jinyou. Density Functional Theory Studies on the CO2 Absorption by 1-Ethylamine-3-methylimidazolium Tetrafluoroborate† [J]. Chem. J. Chinese Universities, 2016, 37(9): 1660. |
| [13] | MA Changmin, LIU Tingyu, CHANG Qiuxiang, LUO Guoyin. Theoretical Studies on the Intrinsic Defects in ZnO and ZnS Crystal† [J]. Chem. J. Chinese Universities, 2016, 37(5): 932. |
| [14] | YANG Bingxing, YE Liping, GU Huijie, XU Huasheng, LUO Yong, LI Huiying. Theoretical Studies on the Structure and Adsorption Properties of Isomorphously Substituted FAU Zeolite† [J]. Chem. J. Chinese Universities, 2016, 37(11): 2018. |
| [15] | PENG Bin, LUO Qiong, LI Nan, ZHANG Xiuhui, LI Qianshu. Theoretical Studies on Mononuclear and Binuclear Osmium Fluoroborylene Carbonyls Os(BF)(CO)n(n=4, 3) and Os2(BF)2(CO)n(n=7, 6, 5, 4)† [J]. Chem. J. Chinese Universities, 2015, 36(11): 2241. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||