

Chem. J. Chinese Universities ›› 2020, Vol. 41 ›› Issue (2): 341.doi: 10.7503/cjcu20190413
• Physical Chemistry • Previous Articles Next Articles
CAO Hongyu1,*( ),MA Zihui2,ZHANG Wenqiong2,TANG Qian1,LI Ruyu2,ZHENG Xuefang1,2,*()
),MA Zihui2,ZHANG Wenqiong2,TANG Qian1,LI Ruyu2,ZHENG Xuefang1,2,*()
Received:2019-07-24
Online:2020-02-10
Published:2019-12-10
Contact:
Hongyu CAO,Xuefang ZHENG
E-mail:caohongyu@foxmail.com;dlxfzheng@126.com
Supported by:CLC Number:
TrendMD:
CAO Hongyu,MA Zihui,ZHANG Wenqiong,TANG Qian,LI Ruyu,ZHENG Xuefang. Structures and Electronic Absorption Spectra of N/Neo-Confused, Doubly N-Confused and Neo-Confused N-Confused Porphyrin Isomers †[J]. Chem. J. Chinese Universities, 2020, 41(2): 341.
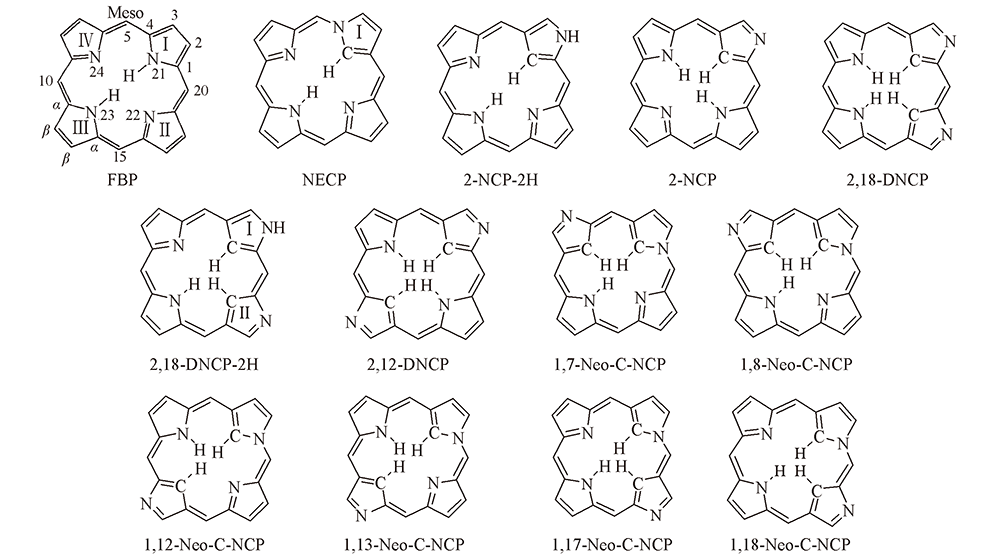
Fig.1 Molecular structures of porphyrin and its isomers
| Compound | 10-30μ/(C·m) | |||
|---|---|---|---|---|
| Vacuum | Benzene | Chloroform | Water | |
| FBP | 0 | 0 | 0 | 0 |
| NECP | 4.27 | 5.50 | 6.37 | 7.47 |
| 2-NCP-2H | 10.88 | 13.78 | 15.85 | 18.58 |
| 2-NCP | 10.51 | 12.88 | 14.51 | 16.65 |
| 2,18-DNCP | 28.52 | 35.86 | 41.20 | 48.21 |
| 2,18-DNCP-2H | 10.44 | 13.04 | 14.85 | 17.15 |
| 2,12-DNCP | 1.63 | 1.93 | 2.07 | 2.07 |
| 1,7-Neo-C-NCP | 12.78 | 15.58 | 17.48 | 19.88 |
| 1,8-Neo-C-NCP | 14.21 | 17.65 | 20.12 | 23.32 |
| 1,12-Neo-C-NCP | 17.68 | 22.15 | 25.32 | 29.42 |
| 1,13-Neo-C-NCP | 18.51 | 23.29 | 26.65 | 31.16 |
| 1,17-Neo-C-NCP | 8.77 | 10.81 | 12.28 | 14.24 |
| 1,18-Neo-C-NCP | 5.77 | 6.61 | 7.07 | 7.51 |
| Compound | 10-30μ/(C·m) | |||
|---|---|---|---|---|
| Vacuum | Benzene | Chloroform | Water | |
| FBP | 0 | 0 | 0 | 0 |
| NECP | 4.27 | 5.50 | 6.37 | 7.47 |
| 2-NCP-2H | 10.88 | 13.78 | 15.85 | 18.58 |
| 2-NCP | 10.51 | 12.88 | 14.51 | 16.65 |
| 2,18-DNCP | 28.52 | 35.86 | 41.20 | 48.21 |
| 2,18-DNCP-2H | 10.44 | 13.04 | 14.85 | 17.15 |
| 2,12-DNCP | 1.63 | 1.93 | 2.07 | 2.07 |
| 1,7-Neo-C-NCP | 12.78 | 15.58 | 17.48 | 19.88 |
| 1,8-Neo-C-NCP | 14.21 | 17.65 | 20.12 | 23.32 |
| 1,12-Neo-C-NCP | 17.68 | 22.15 | 25.32 | 29.42 |
| 1,13-Neo-C-NCP | 18.51 | 23.29 | 26.65 | 31.16 |
| 1,17-Neo-C-NCP | 8.77 | 10.81 | 12.28 | 14.24 |
| 1,18-Neo-C-NCP | 5.77 | 6.61 | 7.07 | 7.51 |
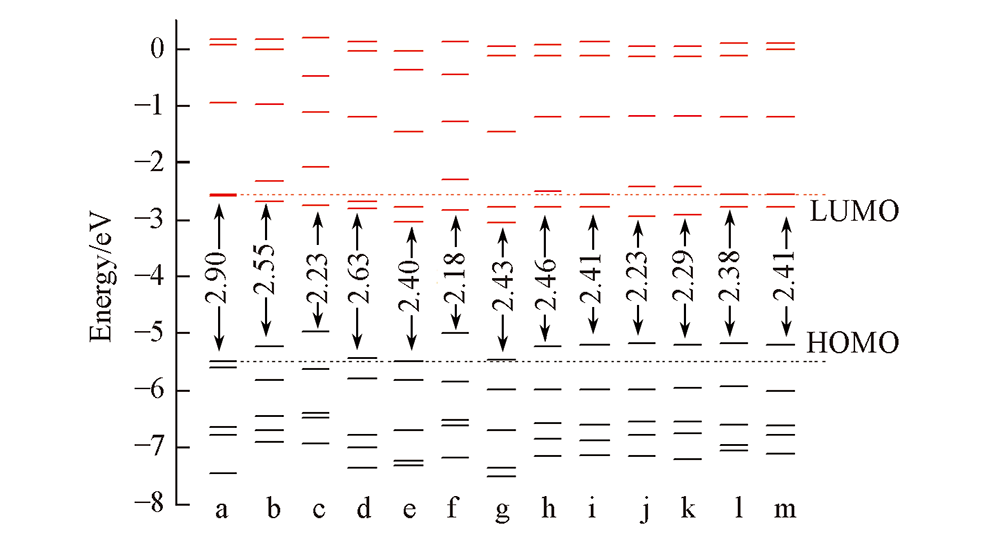
Fig.2 Orbital energy levels and the Egap of porphyrin and its isomers a. FBP; b. NECP; c. 2-NCP-2H; d. 2-NCP; e. 2,18-DNCP; f. 2,18-DNCP-2H; g. 2,12-DNCP; h. 1,7-Neo-C-NCP; i. 1,8-Neo-C-NCP; j. 1,12-Neo-C-NCP; k. 1,13-Neo-C-NCP; l. 1,17-Neo-C-NCP; m. 1,18-Neo-C-NCP.
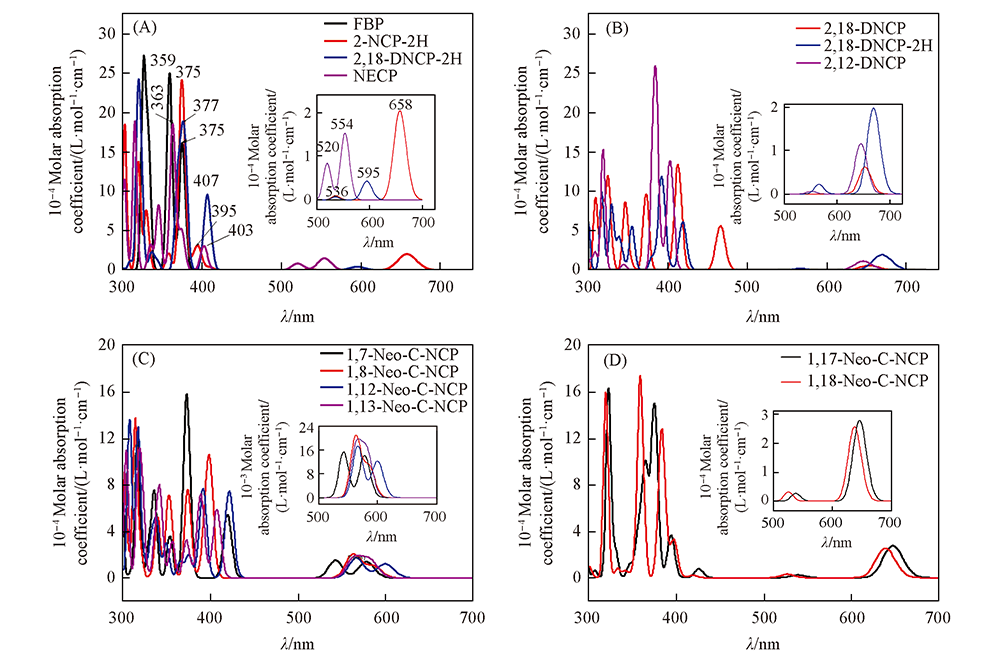
Fig.3 Simulated absorption spectra of porphyrins in vacuum
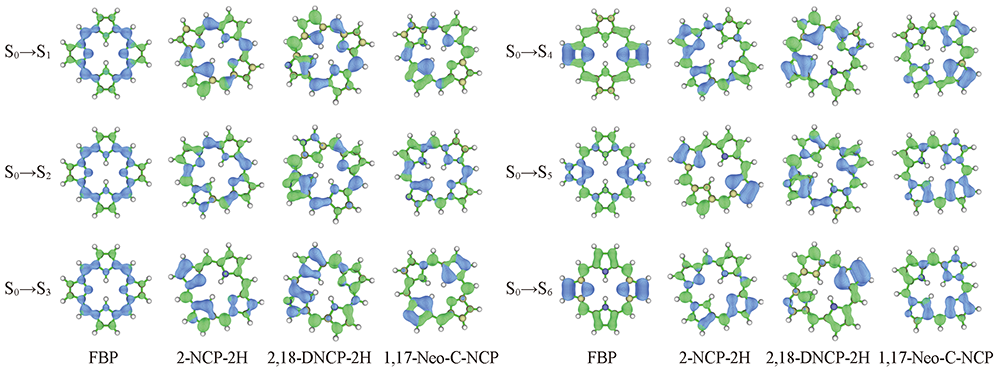
Fig.4 Hole-electron distributions of S1→S6 states in FBP, 2-NCP-2H, 2,18-DNCP-2H and 1,17-Neo-C-NCP The colors of blue and green indicate hole and electron distribution, respectively.
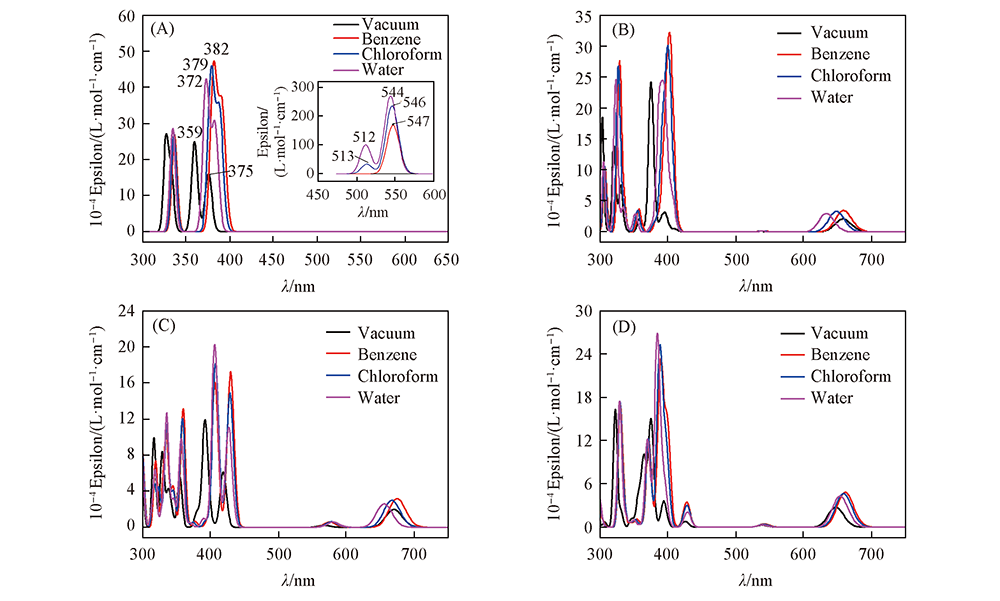
Fig.5 Simulated absorption spectra of FBP(A), 2-NCP-2H(B) , 2,18-DNCP-2H(C) and 1,17-Neo-C-NCP(D) in vacuum, benzene, chloroform and water
| [1] |
Waluk J., Chem. Rev., 2017,117(4), 2447— 2480
doi: 10.1021/acs.chemrev.6b00328 URL |
| [2] |
Liu T. F., Feng D. W., Chen Y. P., Zou L. F., Bosch M., Yuan S., Wei Z. W., Fordham S., Wang K. C., Zhou H. C., J. Am. Chem. Soc., 2015,137(1), 413— 419
doi: 10.1021/ja5111317 URL |
| [3] |
Mathew S., Yella A., Gao P., Humphry-Baker R., Curchod B., Ashari-Astani N., Tavernelli I., Rothlisberger U., Nazeeruddin M. K., Gratzel M., Nature Chem., 2014,6(3), 242— 247
doi: 10.1038/nchem.1861 URL |
| [4] | Kang S. H., Jeong M. J., Eom Y. K., Choi I. T., Kwon S. M., Yoo Y., Kim J., Kwon J., Park J. H., Kim H. K., Adv. Energy Mater., 2017,7(7), 1602117 |
| [5] | Kang H. M., Wang H. Q., Wang H. Y., Wu L. X., Qiu Y. Q., Chem.J. Chinese Universities, 2019,40(5), 965— 972 |
| ( 康慧敏, 王洪强, 王慧莹, 吴黎歆, 仇永清 . 高等学校化学学报, 2019,40(5), 965— 972) | |
| [6] |
Abrahamse H., Hamblin M. R ., Biochem. J., 2016,473(4), 347— 364
doi: 10.1042/BJ20150942 URL |
| [7] | Fukui N., Kim T., Kim D., Osuka A ., J. Am. Chem. Soc., 2017,139(26), 9075— 9088 |
| [8] | Wang S. H., Shang L., Li L. L., Yu Y. J., Chi C. W., Wang K., Zhang J., Shi R., Shen H. Y., Waterhouse G., Liu S. J., Tian J., Zhang T. R., Liu H. Y., Adv. Mater., 2016,28(38), 8379— 8387 |
| [9] | Sun K. F., Cai C., Hou Z. S., Wang Y., Ren Q. Z., Chem. J. Chinese Universities, 2017,38(7), 1117— 1124 |
| ( 孙凯芳, 蔡诚, 侯宗胜, 王颖, 任奇志 . 高等学校化学学报, 2017,38(7), 1117— 1124) | |
| [10] | Kelly J. F., Snell M. E., World J. Urol., 1976,115(2), 150— 151 |
| [11] | Huang P., Qian X. Q., Chen Y., Yu L. D., Lin H., Wane L. Y., Zhu Y. F., Shi J. L., J. Am. Chem. Soc., 2017,139(3), 1275— 1284 |
| [12] | Feng X. L., Shi Y., Xie L. F., Zhang K., Wang X. B., Liu Q. H., Wang P., Med. Chem., 2018,61(16), 7189— 7201 |
| [13] | Li M. L., Peng X. J., Acta Chimica Sinica, 2016,74(12), 959— 968 |
| ( 李明乐, 彭孝军 . 化学学报, 2016,74(12), 959— 968) | |
| [14] | McKenzie L. K., Sazanovich I. V., Baggaley E., Bonneau M., Guerchais V., Williams J., Weinstein J., Bryant H. E., Chem. Eur. J., 2018,23(2), 234— 238 |
| [15] | Vogel E., Schmickler H., Lex J ., Angew., 1986,25(3), 257— 259 |
| [16] | Sessler J. L., Brucker E. A., Weghorn S. J., Angew., 1994,33(22), 2308— 2312 |
| [17] | Callot H. J., Rohrer A., Tschamber T., Metz B., New J. Chem., 1995,19(2), 155— 159 |
| [18] | Vogel E., Bröring M., Erben C., Demuth R., Lex J., Nendel M., Houk K. N., Angew. Chem. Int. Ed., 1997,36(4), 353— 357 |
| [19] | Furuta H., Asano T., Ogawa T ., J. Am. Chem. Soc., 1994,116(2), 767— 768 |
| [20] | Chmielewski P. J., Latos-Grazyński L., Rachlewicz K., Angew. Chem. Int. Ed., 1994,33(7), 779— 781 |
| [21] |
Fujino K., Hirata Y., Kawabe Y., Morimoto T., Srinivasan A., Toganoh M., Miseki Y., Kudo A., Furuta H., . Angew. Chem. Int. Ed., 2011,50(30), 6855— 6859
doi: 10.1002/anie.201100429 URL |
| [22] |
Furuta H., Maeda H., Osuka A ., J. Am. Chem. Soc., 2000,122(5), 803— 807
doi: 10.1021/ja992679g URL |
| [23] | Maeda H., Osuka A., Furuta H ., J. Am. Chem. Soc., 2003,125(51), 15690— 15691 |
| [24] | AbuSalim D. I., Lash T. D., J. Phys. Chem., 2015,119(46), 11440— 11453 |
| [25] | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J., Gaussian 0., Revision D.0., Gaussian Inc., Wallingford CT, 2009 |
| [26] | Scalmani G., Frisch M. J., J. Chem. Phys., 2010,132, 114110 |
| [27] | Bauernschmitt R., Ahlrichs R., Chem. Phys. Lett., 1996,256(4/5), 454— 464 |
| [28] | Casida M. E., Jamorski C., Casida K. C., Salahub D. R., J. Chem. Phys., 1998,108(11), 4439— 4449 |
| [29] | Lu T., Chen F. W., Comp. Chem., 2012,33(5), 580— 592 |
| [30] | Lu T., Chen F. W., J. Mol. Graph. Model, 2012,38, 314— 323 |
| [31] |
Lash T. D., Lammer A. D., Ferrence G. M., Angew. Chem. Int. Ed., 2011,50(41), 9718— 9721
doi: 10.1002/anie.201104826 URL |
| [32] | Furuta H., Ishizuka T., Osuka A., Dejima H., Nakagawa H., Ishikawa Y ., J. Am. Chem. Soc., 2001,123(25), 6207— 6208 |
| [33] | Liu X. J., Pan H. Q., Meng J., Mol. Struct., Theochem., 2006,765(1), 61— 69 |
| [34] | Cao H. Y., Hao J. Y., Si D. H., Tang Q., Zheng X. F., Hao C., Chin. J. Struct. Chem., 2018,37(8), 1223— 1232 |
| [35] | Edwards L., Dolphin D. H., Gouterman M., Adler A. D., Mol. Spectrosc., 1971,38(1), 16— 32 |
| [36] | Tanaka T., Osuka A., Chem. Rev., 2017,177(4), 2584— 2640 |
| [37] | Petit L., Quartrolo A., Admo C., Russo N ., J. Phys. Chem. B, 2006,110(5), 2398— 2404 |
| [1] | HE Hongrui, XIA Wensheng, ZHANG Qinghong, WAN Huilin. Density-functional Theoretical Study on the Interaction of Indium Oxyhydroxide Clusters with Carbon Dioxide and Methane [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220196. |
| [2] | QIU Liqi, YAO Xiangyang, HE Liangnian. Visible-light-driven Selective Reduction of Carbon Dioxide Catalyzed by Earth-abundant Metalloporphyrin Complexes [J]. Chem. J. Chinese Universities, 2022, 43(7): 20220064. |
| [3] | YU Bin, CHEN Xiaoyan, ZHAO Yue, CHEN Weichang, XIAO Xinyan, LIU Haiyang. Graphene Oxide-based Cobalt Porphyrin Composites for Electrocatalytic Hydrogen Evolution Reaction [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210549. |
| [4] | WANG Yuanyue, AN Suosuo, ZHENG Xuming, ZHAO Yanying. Spectroscopic and Theoretical Studies on 5-Mercapto-1,3,4-thiadiazole-2-thione Microsolvation Clusters [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220354. |
| [5] | YING Fuming, JI Chenru, SU Peifeng, WU Wei. λ-DFCAS: A Hybrid Density Functional Complete Active Space Self Consistent Field Method [J]. Chem. J. Chinese Universities, 2021, 42(7): 2218. |
| [6] | ZHU Qichen, XIONG Ming, TAO Siyu, TANG Siwei, REN Qizhi. Effect of Light Source on the Photocatalytic Performance of Dihydroxynaphthalene by Water-soluble Sulfonated Porphyrins [J]. Chem. J. Chinese Universities, 2021, 42(6): 1933. |
| [7] | MA Zihui, WANG Mengyan, CAO Hongyu, TANG Qian, WANG Lihao, ZHENG Xuefang. Transient Absorption and Decay Kinetic Properties of Photo-excited Metal Coordinated Tetraphenylporphyrin [J]. Chem. J. Chinese Universities, 2021, 42(3): 767. |
| [8] | XIE Xingyu, ZHAO Yaxiang, ZHAO Lizhi, LI Rishun, WU Dihao, YE Hui, XIN Qingping, LI Hong, ZHANG Yuzhong. Colorimetric Detection Method for H2O2 Based on Two-dimensional Metal-organic Frameworks of Metalloporphyrin [J]. Chem. J. Chinese Universities, 2020, 41(8): 1776. |
| [9] | WEI Xin, DENG Yaoliang, ZHENG Xuming, ZHAO Yanying. Ground Structure and Excited State Proton Transfer Reaction of 2-Aminobenzothiazole [J]. Chem. J. Chinese Universities, 2019, 40(8): 1679. |
| [10] | ZHI Shasha,BAN Ying,XU Zhiguang,XU Xuan. Electron Transport of Metal String Complexes of [MM'M″(dpa)4(Cl)2](M=Co, Ni; M',M″=Co, Rh)† [J]. Chem. J. Chinese Universities, 2019, 40(5): 980. |
| [11] | LIU Qiuna,XU Wenwen,LIU Maozhu,WANG Huigang,ZHENG Xuming. Study on Raman Spectroscopy Non-coincidence Effect of Propionic Anhydride C=O Vibration Mode† [J]. Chem. J. Chinese Universities, 2019, 40(5): 932. |
| [12] |
ZHOU Xiaofeng,ZHOU Yanbing,TANG Chunmei.
Hydrogen Storage Capacity of the Alkaline Earth Metal Mg Exohedral Doped Boron Cage B40M |
| [13] | ZHOU Hegen,JIN Hua,GUO Huirui,LIN Jing,ZHANG Yongfan. Electronic Structures and Optical Properties of Cu-based Semiconductors with Chalcopyrite-type Structure† [J]. Chem. J. Chinese Universities, 2019, 40(3): 518. |
| [14] | YANG Yejin,YOU Jinglin,WANG Jian,WANG Min,HE Yingxia,WU Zhidong. In-situ High Temperature Raman Spectroscopic and Decomposition Thermodynamic Study of the Structure of Potassium Hydrogen Sulfate and Its Melt† [J]. Chem. J. Chinese Universities, 2018, 39(10): 2272. |
| [15] | HE Wenli, CHEN Meng, QIAN Dongjin. Pd(Ⅱ)-Directed Layer-by-layer Assembly of Multiporphyrin Arrays on Silica Nanoparticle Surfaces for Photocurrent Generation and Photochromism of Viologens† [J]. Chem. J. Chinese Universities, 2017, 38(9): 1654. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||