

Chem. J. Chinese Universities ›› 2023, Vol. 44 ›› Issue (11): 20230276.doi: 10.7503/cjcu20230276
• Physical Chemistry • Previous Articles Next Articles
ZHOU Zihao, WANG Sihao, HUANG Daichuan, LIU Bo, NING Hongbo( )
)
Received:2023-06-10
Online:2023-11-10
Published:2023-09-04
Contact:
NING Hongbo
E-mail:hbning@swjtu.edu.cn
Supported by:CLC Number:
TrendMD:
ZHOU Zihao, WANG Sihao, HUANG Daichuan, LIU Bo, NING Hongbo. Molecular Dynamics Simulation Study on High Temperature Oxidation Mechanism of n-Propylbenzene[J]. Chem. J. Chinese Universities, 2023, 44(11): 20230276.
| Equivalence ratio(ϕ) | n(n⁃Propylbenzene)/n(Oxygen) | T/K | Density/(g·cm-3) | Ensemble |
|---|---|---|---|---|
| 1.0 | 50/600 | 2900 | 0.35 | NVT |
| 0.5 | 10/240 | 2300—3500 | 0.05—0.35 | NVT |
| 1.0 | 10/120 | 2300—3500 | 0.05—0.35 | NVT |
| 2.0 | 10/60 | 2300—3500 | 0.05—0.35 | NVT |
Table 1 Temperature, density, and equivalence ratio of n-propylbenzene oxidation system
| Equivalence ratio(ϕ) | n(n⁃Propylbenzene)/n(Oxygen) | T/K | Density/(g·cm-3) | Ensemble |
|---|---|---|---|---|
| 1.0 | 50/600 | 2900 | 0.35 | NVT |
| 0.5 | 10/240 | 2300—3500 | 0.05—0.35 | NVT |
| 1.0 | 10/120 | 2300—3500 | 0.05—0.35 | NVT |
| 2.0 | 10/60 | 2300—3500 | 0.05—0.35 | NVT |
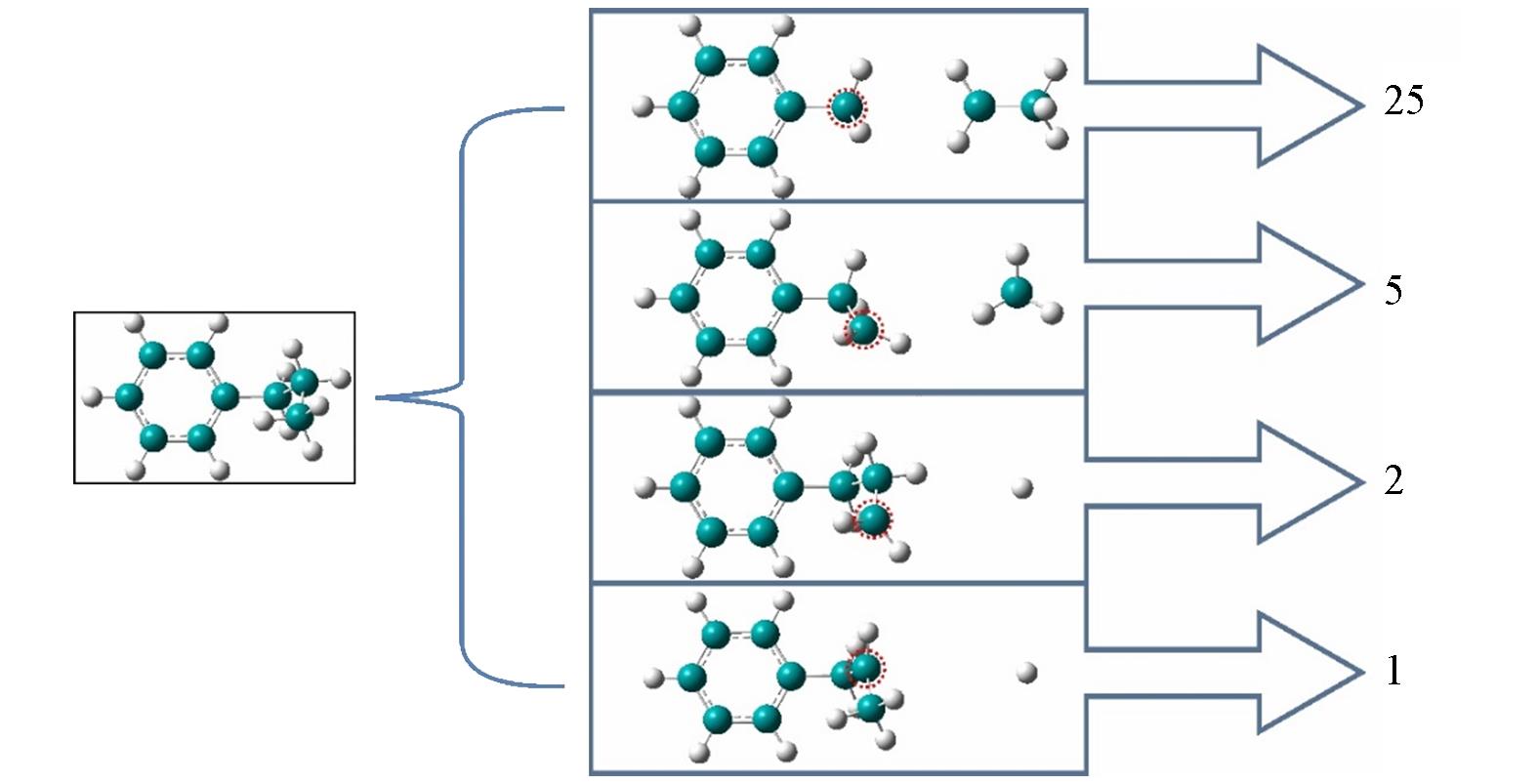
| Reaction | Ebond/(kJ·mol-1) | ||
|---|---|---|---|
| B3LYP | ReaxFF | Ref. | |
| C6H5CH2CH2CH3→C6H5CH2CH2CH2·+H· | 424.19 | 444.38 | 420.09[ |
| C6H5CH2CH2CH3→C6H5CH2CH·CH3+H· | 407.55 | 435.89 | 408.80[ |
| C6H5CH2CH2CH3→C6H5CH·CH2CH3+H· | 359.52 | 381.68 | 366.17[ |
| C6H5CH2CH2CH3→C6H5CH2CH2·+CH3· | 358.02 | 372.39 | 371.60[ |
| C6H5CH2CH2CH3→C6H5CH2·+CH3CH2· | 290.30 | 298.74 | 322.28[ |
| C6H5CH2CH2CH3→C6H5·+CH3CH2CH2· | 400.32 | 424.86 | 421.34[ |
Table 2 Calculated bond dissociation energies of n-propylbenzene using B3LYP and ReaxFF methods and the corresponding literature results
| Reaction | Ebond/(kJ·mol-1) | ||
|---|---|---|---|
| B3LYP | ReaxFF | Ref. | |
| C6H5CH2CH2CH3→C6H5CH2CH2CH2·+H· | 424.19 | 444.38 | 420.09[ |
| C6H5CH2CH2CH3→C6H5CH2CH·CH3+H· | 407.55 | 435.89 | 408.80[ |
| C6H5CH2CH2CH3→C6H5CH·CH2CH3+H· | 359.52 | 381.68 | 366.17[ |
| C6H5CH2CH2CH3→C6H5CH2CH2·+CH3· | 358.02 | 372.39 | 371.60[ |
| C6H5CH2CH2CH3→C6H5CH2·+CH3CH2· | 290.30 | 298.74 | 322.28[ |
| C6H5CH2CH2CH3→C6H5·+CH3CH2CH2· | 400.32 | 424.86 | 421.34[ |
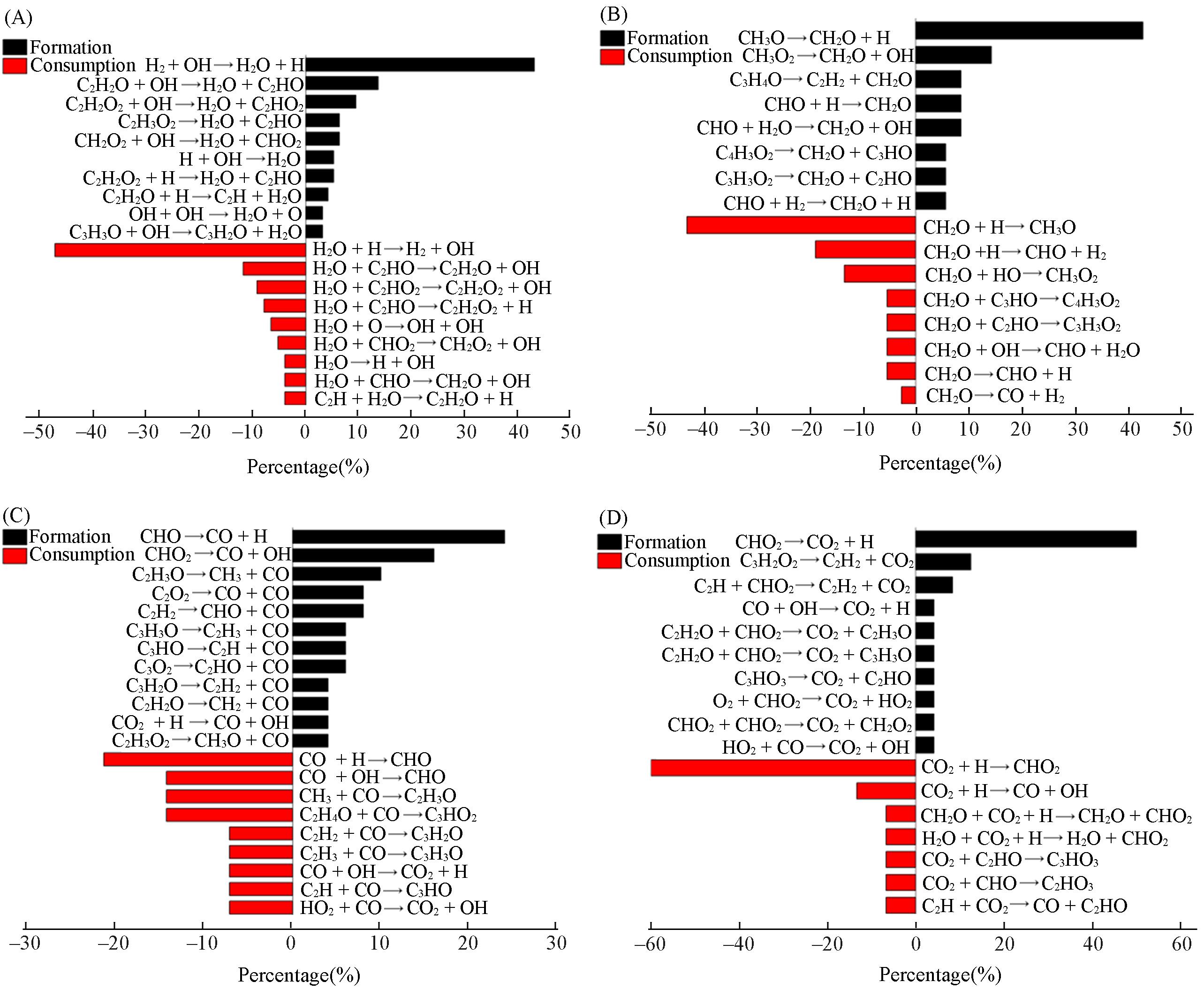
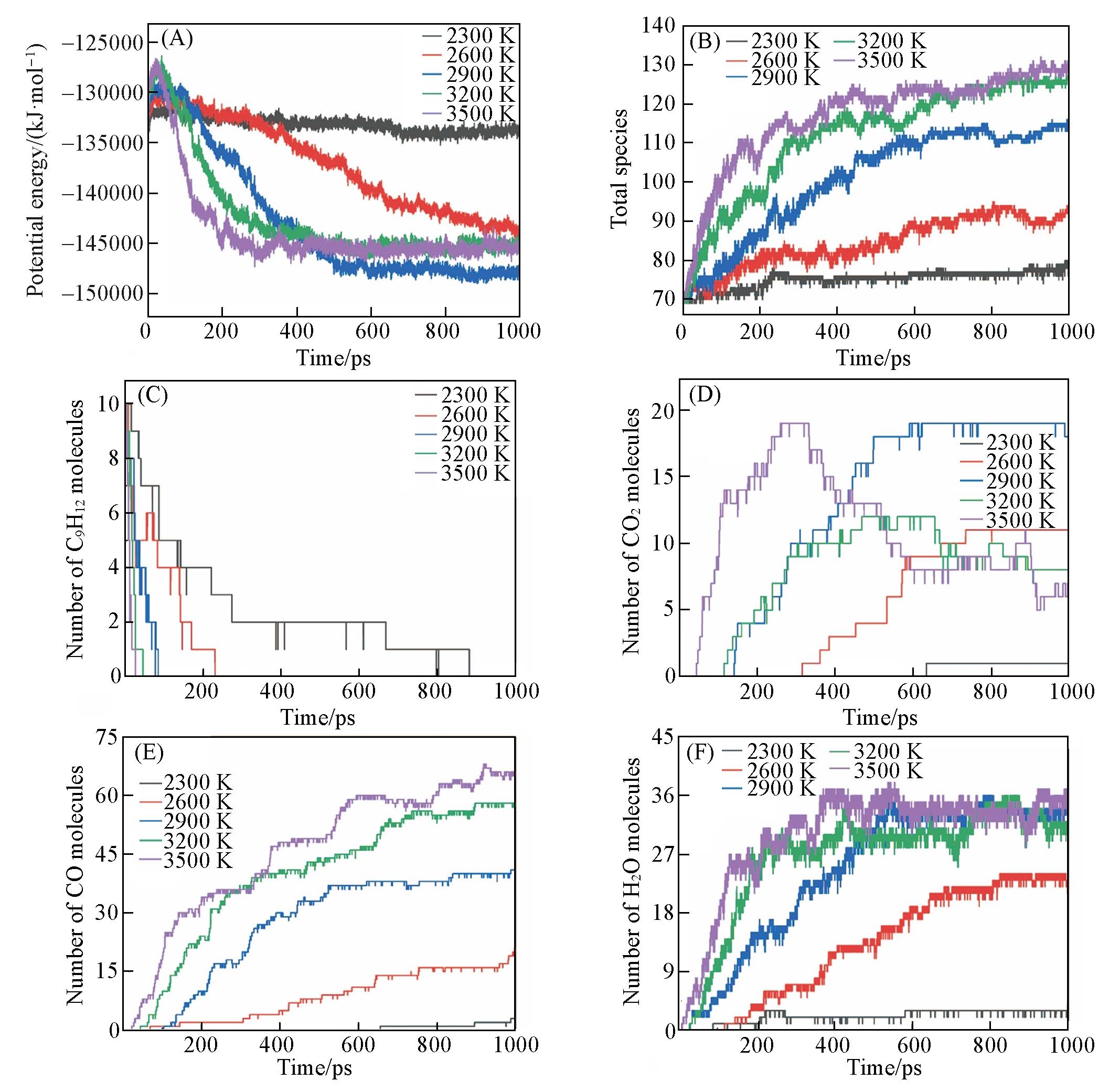
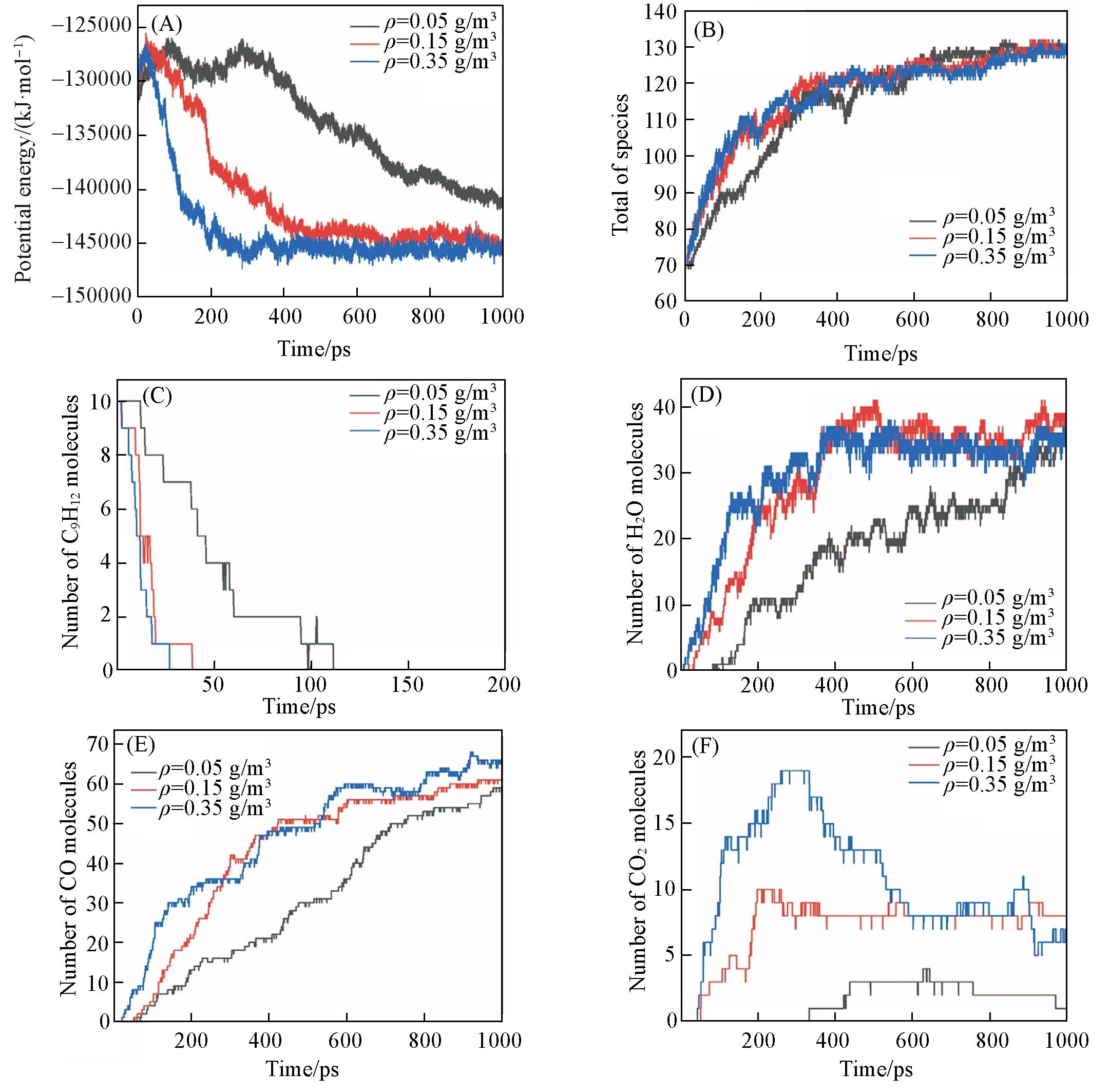
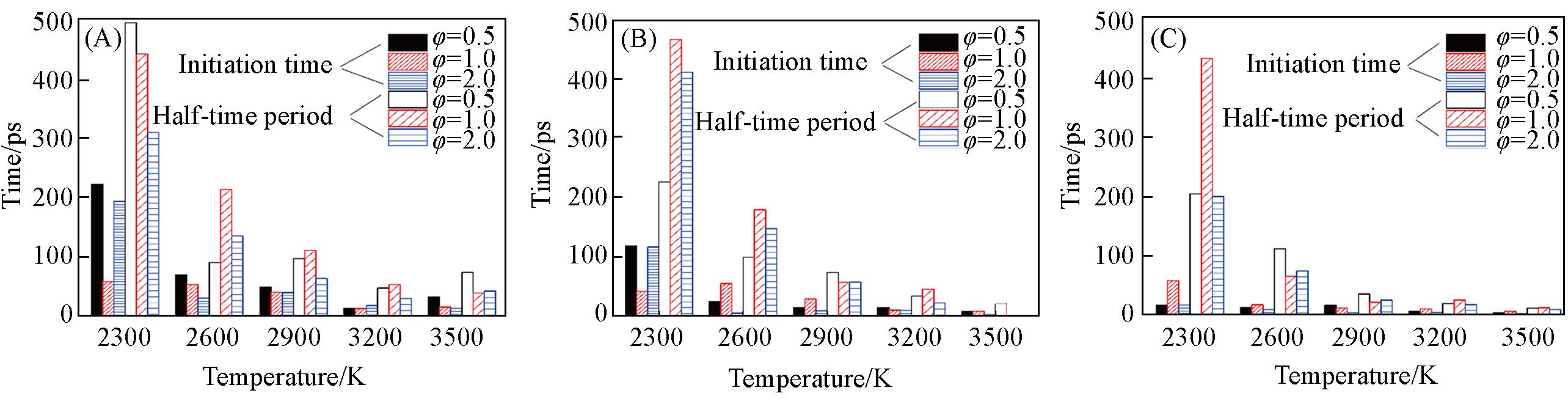
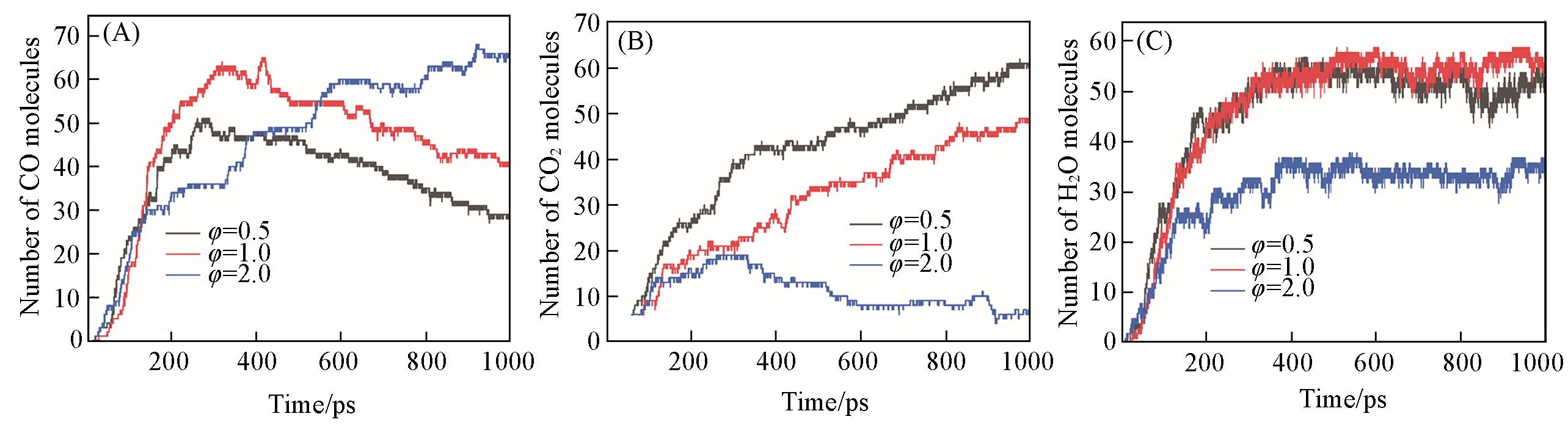
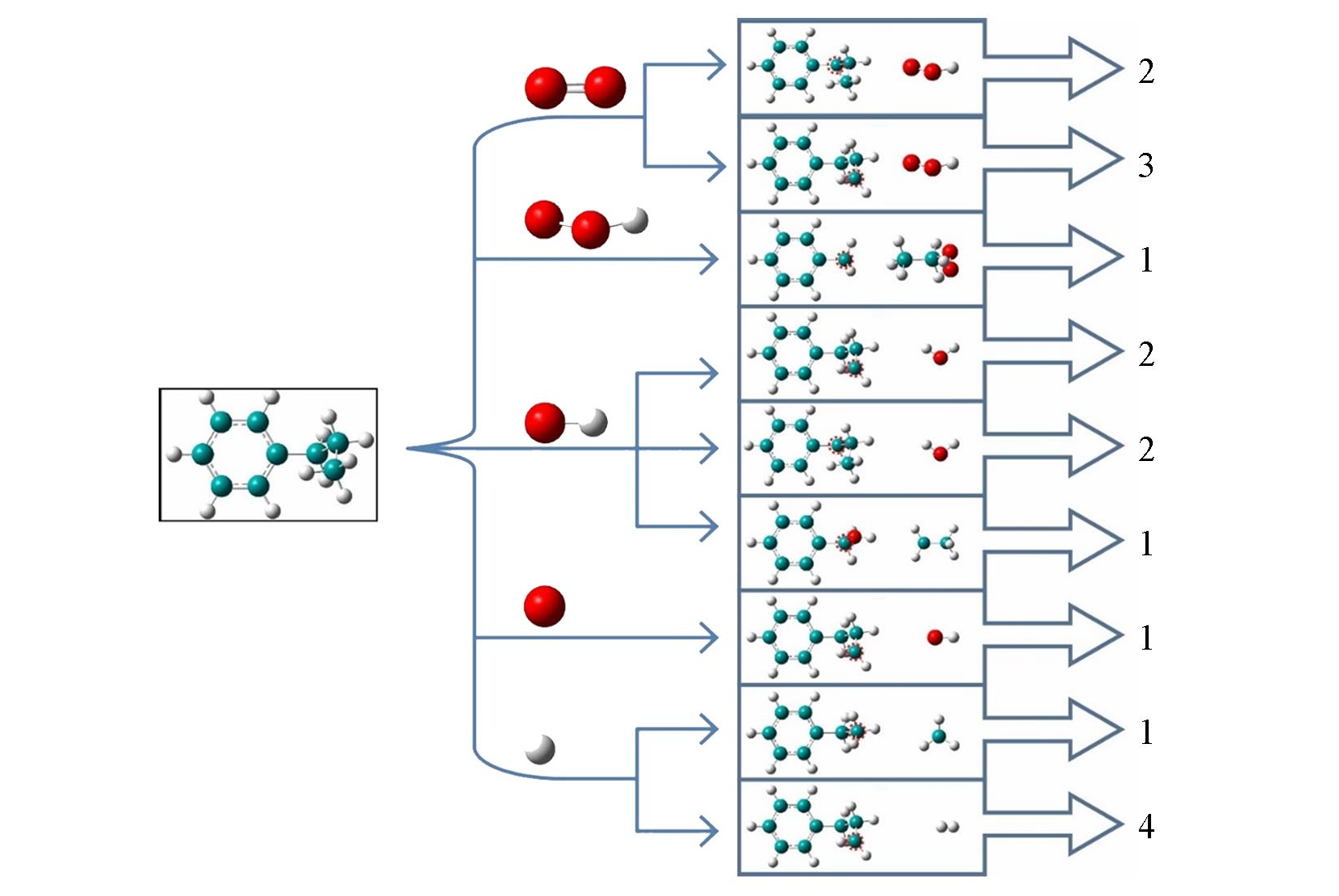
| Method | Density/(g·cm-3) | Equivalence ratio | Ea/(kJ·mol-1) | A/(cm3·mol-1·s-1) |
|---|---|---|---|---|
| ReaxFF | 0.05 | 0.5 | 129.66 | 0.66×109 |
| 1.0 | 135.98 | 0.91×109 | ||
| 2.0 | 129.08 | 0.90×109 | ||
| 0.15 | 0.5 | 135.52 | 1.72×109 | |
| 1.0 | 172.43 | 5.24×109 | ||
| 2.0 | 205.28 | 2.65×1010 | ||
| 0.35 | 0.5 | 183.17 | 1.67×1010 | |
| 1.0 | 180.49 | 1.48×1010 | ||
| 2.0 | 142.04 | 0.38×109 | ||
| Expt.[ | — | — | 138.02 | 6.04×1010 |
Table 3 Fitted Arrhenius parameters including Ea and A
| Method | Density/(g·cm-3) | Equivalence ratio | Ea/(kJ·mol-1) | A/(cm3·mol-1·s-1) |
|---|---|---|---|---|
| ReaxFF | 0.05 | 0.5 | 129.66 | 0.66×109 |
| 1.0 | 135.98 | 0.91×109 | ||
| 2.0 | 129.08 | 0.90×109 | ||
| 0.15 | 0.5 | 135.52 | 1.72×109 | |
| 1.0 | 172.43 | 5.24×109 | ||
| 2.0 | 205.28 | 2.65×1010 | ||
| 0.35 | 0.5 | 183.17 | 1.67×1010 | |
| 1.0 | 180.49 | 1.48×1010 | ||
| 2.0 | 142.04 | 0.38×109 | ||
| Expt.[ | — | — | 138.02 | 6.04×1010 |
| 1 | Liu Y. X., Wang B. Y., Weng J. J., Yu D., Richter S., Kick T., Tian Z. Y., Combust. Flame, 2018, 191, 53—65 |
| 2 | Dagaut P., El Bakali A., Ristori A., Fuel, 2006, 85(7/8), 944—956 |
| 3 | Xu J. Q., Guo J. J., Liu A. K., Wang J. L., Tan N. X., Li X. Y., Acta Phys. Chim. Sin., 2015, 31(4), 643—652 |
| 徐佳琪, 郭俊江, 刘爱科, 王健礼, 谈宁馨, 李象远. 物理化学学报, 2015, 31(4), 643—652 | |
| 4 | Dooley S., Won S. H., Heyne J., Farouk T. I., Ju Y., Dryer F. L., Brezinsky K., Combust. Flame, 2012, 159(4), 1444—1466 |
| 5 | Johnston R. J., Farrell J. T., Proc. Combust. Inst., 2005, 30(1), 217—224 |
| 6 | Hui X., Das A. K., Kumar K., Sung C. J., Dooley S., Dryer F. L., Fuel, 2012, 97, 695—702 |
| 7 | Hui X., Sung C. J., Fuel, 2013, 109, 191—200 |
| 8 | Ji C., Dames E., Wang H., Egolfopoulos F. N., Combust. Flame, 2012, 159(3), 1070—1081 |
| 9 | Mehl M., Herbinet O., Dirrenberger P., Bounaceur R., Glaude P. A., Battin⁃Leclerc F., Pitz W. J., Proc. Combust. Inst., 2015, 35(1), 341—348 |
| 10 | Roubaud A., Minetti R., Sochet L. R.., Combust. Flame, 2000, 121(3), 535—541 |
| 11 | Darcy D., Mehl M., Simmie J. M., Würmel J., Metcalfe W. K., Westbrook C. K., Curran H. J., Proc. Combust. Inst., 2013, 34(1), 411—418 |
| 12 | Darcy D., Nakamura H., Tobin C. J., Mehl M., Metcalfe W. K., Pitz W. J., Curran H. J., Combust. Flame, 2014, 161(1), 65—74 |
| 13 | Darcy D., Nakamura H., Tobin C. J., Mehl M., Metcalfe W. K., Pitz W. J., Curran H. J., Combust. Flame, 2014, 161(6), 1460—1473 |
| 14 | Gudiyella S., Brezinsky K., Combust. Flame, 2012, 159(3), 940—958 |
| 15 | Gudiyella S., Brezinsky K., Proc. Combust. Inst., 2013, 34(1), 1767—1774 |
| 16 | Anderson H., McEnally C. S., Pfefferle L. D., Proc. Combust. Inst., 2000, 28(2), 2577—2583 |
| 17 | Wang Z., Li Y., Zhang F., Zhang L., Yuan W., Wang Y., Qi F., Proc. Combust. Inst., 2013, 34(1), 1785—1793 |
| 18 | Li Y., Cai J., Zhang L., Yang J., Wang Z., Qi F., Proc. Combust. Inst., 2011, 33(1), 617—624 |
| 19 | Darcy D., Tobin C. J., Yasunaga K., Simmie J. M., Würmel J., Metcalfe W. K., Tidjani N., Ahmed S. S., Westbrook C. K., Curran H. J., Combust. Flame, 2012, 159(7), 2219—2232 |
| 20 | Robinson R. K., Lindstedt R. P., Combust. Flame, 2013, 160(12), 2642—2653 |
| 21 | Altarawneh M., Dlugogorski B. Z., Combust. Flame, 2015, 162(4), 1406—1416 |
| 22 | Zhou C. W., Simmie J. M., Somers K. P., Goldsmith C. F., Curran H. J., J. Phys. Chem. A, 2017, 121(9), 1890—1899 |
| 23 | Shang Y., Ning H., Shi J., Luo S. N., Chem. Res. Chinese Universities, 2021, 37(3), 711—717 |
| 24 | van Duin A. C. T., Dasgupta S., Lorant F., Goddard W. A., J. Phys. Chem. A, 2001, 105(41), 9396—9409 |
| 25 | Li X., Zheng M., Ren C., Guo L., Energy Fuels, 2021, 35(15), 11707—11739 |
| 26 | Chenoweth K., van Duin A. C. T., Goddard W. A., J. Phys. Chem. A, 2008, 112(5), 1040—1053 |
| 27 | Chenoweth K., van Duin A. C. T., Dasgupta S., Goddard W. A., J. Phys. Chem. A, 2009, 113(9), 1740—1746 |
| 28 | Wang Q. D., Wang J. B., Li J. Q., Tan N. X., Li X. Y., Combust. Flame, 2011, 158(2), 217—226 |
| 29 | Cheng X. M., Wang Q. D., Li J. Q., Wang J. B., Li X. Y., J. Phys. Chem. A, 2012, 116(40), 9811—9818 |
| 30 | Liu X. L., Li, X. X., Han S., Qiao X. J., Zhong B. J., Guo L., Acta Phys. Chim. Sin., 2016, 32(6), 1424—1433 |
| 刘晓龙, 李晓霞, 韩嵩, 乔显杰, 钟北京, 郭力. 物理化学学报, 2016, 32(6), 1424—1433 | |
| 31 | Liu J. X., Min J., Xu H. J., Ren H. S., Tan N. X., Chem. J. Chinese Universities, 2022, 43(4),20210834 |
| 刘嘉欣, 闵杰, 许华杰, 任海生, 谭宁馨. 高等学校化学学报, 2022, 43(4),20210834 | |
| 32 | Berendsen H. J., Postma J., Gunsteren W., DiNola A. D., Haak J. R., J. Chem. Phys., 1984, 81(8), 3684—3690 |
| 33 | Lindgren E. B., Monteiro J. G. S., dos Santos A. R., Fleming F. P., Barbosa A. G. H., Fuel, 2021, 303, 121205 |
| 34 | So M. R., Rensen., Voter A. F., J. Chem. Phys., 2000, 112(21), 9599—9606 |
| 35 | Ashraf C., van Duin A. C. T., J. Phys. Chem. A, 2017, 121(5), 1051—1068 |
| 36 | Martínez L., Andrade R., Birgin E. G., Martínez J. M., J. Comput. Chem., 2010, 30(13), 2157—2164 |
| 37 | Plimpton S., J. Comput. Phys., 1995, 117(1), 1—19 |
| 38 | Döntgen M., Przybylski⁃Freund M. D., Kröger L. C., Kopp W. A., Ismail A. E., Leonhard K., J. Chem. Theory Comput., 2015, 11(6), 2517—2524 |
| 39 | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams⁃Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J., Gaussian 16., Revision B. 01, Gaussian Inc., Wallingford CT, 2016 |
| 40 | Luo Y. R., Comprehensive Handbook of Chemical Bond Energies, CRC Press, London, 2007 |
| 41 | Kirkpatrick A. T., Internal Combustion Engines: Applied Thermosciences, John Wiley & Sons, Hoboken, 2020 |
| 42 | Liang J., Li F., Cao S., Li X., Jia M. X., Wang Q. D., Fuel, 2022, 325, 124940 |
| 43 | Yuan W., Li Y., Dagaut P., Wang Y., Wang Z., Qi F., Combust. Flame, 2017, 186, 178—192 |
| [1] | XU Jianing, BAI Wenjing, LOU Yuhan, YU Haipeng, DOU Shuo. Electrocatalytic Oxidative Cleavage of Lignin: Facile and Efficient Biomass Valorization Strategy [J]. Chem. J. Chinese Universities, 2023, 44(5): 20220749. |
| [2] | FU Zhongheng, CHEN Xiang, YAO Nan, YU Legeng, SHEN Xin, ZHANG Rui, ZHANG Qiang. Research Advances in Transport Mechanism of Lithium Ions in Solid Electrolytes [J]. Chem. J. Chinese Universities, 2023, 44(5): 20220703. |
| [3] | GAO Fengyu, CHEN Du, LUO Ning, YAO Xiaolong, DUAN Erhong, YI Honghong, ZHAO Shunzheng, TANG Xiaolong. Catalytic Performance and Reaction Mechanism of Chlorobenzene Oxidation over MnO x -CeO2 Catalyst [J]. Chem. J. Chinese Universities, 2023, 44(4): 20220690. |
| [4] | XIA Wenwen, YU Hongjing, WANG Shiye, YAO Li, LI Xiangyuan. Combustion Mechanism Construction Based on Minimized Reaction Network: Combustion of Aromatic Hydrocarbon [J]. Chem. J. Chinese Universities, 2023, 44(4): 20220616. |
| [5] | LI Jichen, CAI Shanshan, PENG Jubo, LI Hongfei, DUAN Xiaozheng. Molecular Dynamics Simulation of Structural Variations of Ionic Polymeric Vesicles under Electric Field [J]. Chem. J. Chinese Universities, 2023, 44(2): 20220553. |
| [6] | SHEN Qi, CHEN Haiyao, GAO Denghui, ZHAO Xi, NA Risong, LIU Jia, HUANG Xuri. Interaction Mechanism of the Natural Product Falcarindiol and Human GABAA Receptor [J]. Chem. J. Chinese Universities, 2023, 44(2): 20220500. |
| [7] | LIAO Shouwei, LIU Yanchang, SHI Zenan, ZHAO Daohui, WEI Yanying, LI Libo. Molecular Dynamics Simulation of Ion Adsorption at Water/Graphene Interface: Force Field Parameter Optimization and Adsorption Mechanism [J]. Chem. J. Chinese Universities, 2023, 44(10): 20230155. |
| [8] | ZHOU Zixuan, YANG Haiyan, SUN Yuhan, GAO Peng. Recent Progress in Heterogeneous Catalysts for the Hydrogenation of Carbon Dioxide to Methanol [J]. Chem. J. Chinese Universities, 2022, 43(7): 20220235. |
| [9] | YANG Dan, LIU Xu, DAI Yihu, ZHU Yan, YANG Yanhui. Research Progress in Electrocatalytic CO2 Reduction Reaction over Gold Clusters [J]. Chem. J. Chinese Universities, 2022, 43(7): 20220198. |
| [10] | GAO Zhiwei, LI Junwei, SHI Sai, FU Qiang, JIA Junru, AN Hailong. Analysis of Gating Characteristics of TRPM8 Channel Based on Molecular Dynamics [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220080. |
| [11] | LIU Jiaxin, MIN Jie, XU Huajie, REN Haisheng, TAN Ningxin. Interaction Between Produced Radicals During Ethylene Combustion and Nitrogen Molecules Based on Reaxff Molecular Dynamics Simulation [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210834. |
| [12] | SUN Cuihong, LYU Liqiang, LIU Ying, WANG Yan, YANG Jing, ZHANG Shaowen. Mechanism and Kinetics on the Reaction of Isopropyl Nitrate with Cl, OH and NO3 Radicals [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210591. |
| [13] | HU Bo, ZHU Haochen. Dielectric Constant of Confined Water in a Bilayer Graphene Oxide Nanosystem [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210614. |
| [14] | ZHANG Mi, TIAN Yafeng, GAO Keli, HOU Hua, WANG Baoshan. Molecular Dynamics Simulation of the Physicochemical Properties of Trifluoromethanesulfonyl Fluoride Dielectrics [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220424. |
| [15] | CHENG Yuanyuan, XI Biying. Theoretical Study on the Fragmentation Mechanism of CH3SSCH3 Radical Cation Initiated by OH Radical [J]. Chem. J. Chinese Universities, 2022, 43(10): 20220271. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||