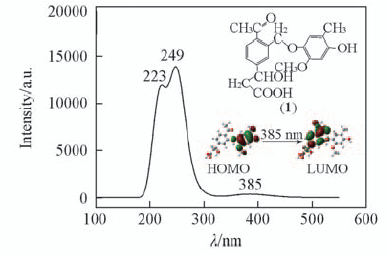
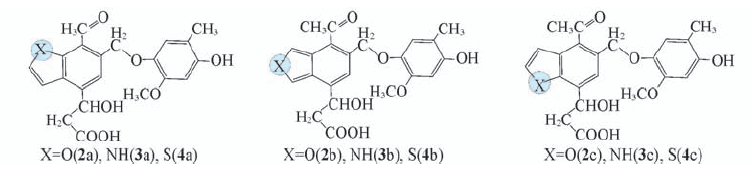
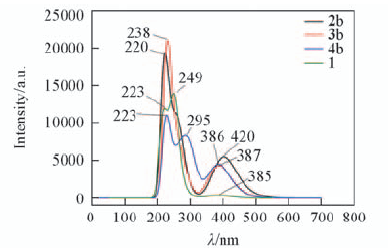
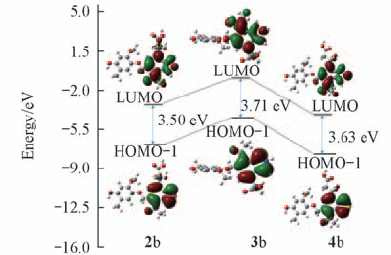
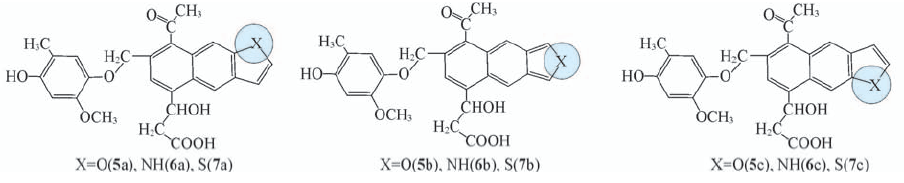
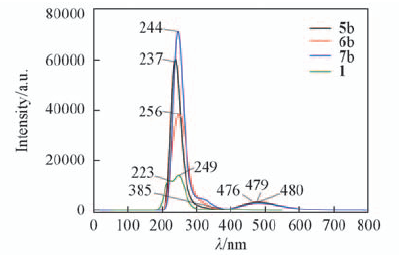
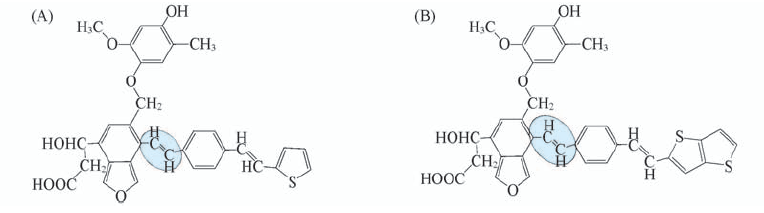
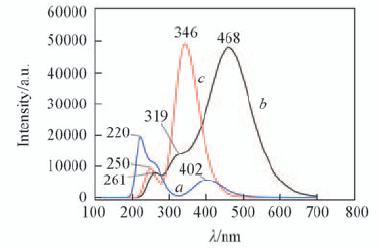
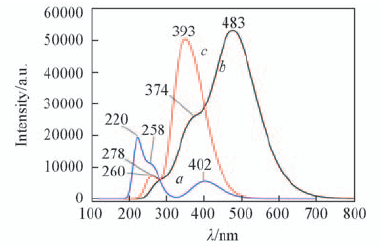
| [1] |
Chen P., The Properties, Classification and Applications of Chinese Coal, Chemical Industry Press, Beijing, 2007, 24—25
|
|
(陈鹏. 国煤炭性质、分类和利用, 北京: 化学工业出版社, 2007, 24—25)
|
| [2] |
Wan Y. Z., Gao J. R., Xiao L., Tao X. X., Liu J. T., Coal Eng., 2010, 4, 75—79
|
|
(万永周, 高俊荣, 肖雷, 陶秀祥, 刘炯天. 煤炭工程, 2010, 4, 75—79)
|
| [3] |
Christian B., Fuels, 2004, 83(3), 267—276
|
| [4] |
Shao J. J., Northwest Coal, 2009, 7(2), 17—22)
|
|
(邵俊杰. 西北煤炭, 2009, 7(2), 17—22)
|
| [5] |
Guo J.J., Theoretical Studies of Structures and Properties of Coals, Xiamen Universities, Xiamen, 2013
|
|
(郭娟娟. 煤结构与性质的理论研究, 厦门: 厦门大学, 2013)
|
| [6] |
Wang J.R., Jin Z. X., Deng C. B., Quantum Chemistry Theory of Coal Spontaneous Combustion, Science Press, Beijing, 2007, 1—5
|
|
(王继仁, 金智新, 邓存宝. 煤自燃量子化学理论, 北京: 科学出版社, 2007, 1—5)
|
| [7] |
Li H., Zhang Y. T., Wang G. H., Zhou A. N., Appl. Chem. Ind., 2007, 36(4), 325—327
|
|
(李慧, 张亚婷, 汪广恒, 周安宁. 应用化工, 2007, 36(4), 325—327)
|
| [8] |
Jiang Y. F., Li K. S., Zhou A. N., Yang Z. Y., Coal Convers., 2004, 27(4), 83—86
|
|
(姜玉凤, 李侃社, 周安宁, 杨致远. 煤炭转化, 2004, 27(4), 83—86)
|
| [9] |
Zhang Y. T., Zhou A. N., J. Xi’an Univ. Sci. Tech., 2008, 2, 344—348
|
|
(张亚婷, 周安宁. 西安科技大学学报, 2008, 2, 344—348 )
|
| [10] |
Carlo A., Denis J., Chem. Soc. Rev., 2013, 42, 845—846
|
| [11] |
Jones R. N., Chem. Rev., 1947, 41(2), 353—371
|
| [12] |
Muranaka A., Yasuike S., Liu C.Y., Kurita J., Kakusawa N., Tsuchiya T., Okuda M., Kobayashi N., Matsumoto Y., Yoshida K., Hashizumi D., Uchiyama M., J. Phys. Chem. A, 2009, 113(2), 464—473
|
| [13] |
Mishra H., Maheshwary S., Tripathi H. B., Sathyamurthy N., J. Phys. Chem. A, 2005, 109, 2746—2754
|
| [14] |
Karabacak M.,Cinar Z., Kurt M., Sudha S., Sundaraganesanc N., Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2012, 85, 179—189
|
| [15] |
Karabacak M., Cinar M., Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2012, 86, 590—599
|
| [16] |
Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., et al., Gaussian 09, Revision B. 01, Gaussian Inc., Wallingford CT, 2009
|
| [17] |
Chen. C. G., Xian X. F., Coal Convers., 1998, 21(2), 7—12
|
|
(陈昌国, 鲜学福. 煤炭转化, 1998, 21(2), 7—12)
|
| [18] |
Wang S.Y., Study of Lignite Structure by Molecular Dynamics Simulation and Quantum Chemistry, Taiyuan University of Technology, Taiyuan, 2004
|
|
(王三跃. 褐煤结构的分子动力学模拟及量子化学研究, 太原: 太原理工大学, 2004)
|
| [19] |
Jia Y., The Experimental Analysis of Brown Coal Structure, Taiyuan University of Technology, Taiyuan, 2002
|
|
(贾燕. 褐煤结构的实验分析, 太原: 太原理工大学, 2002)
|
 )
)