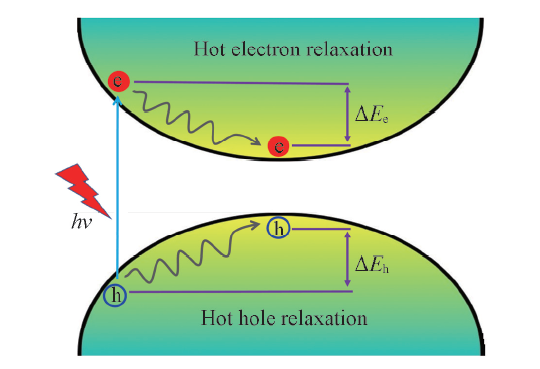
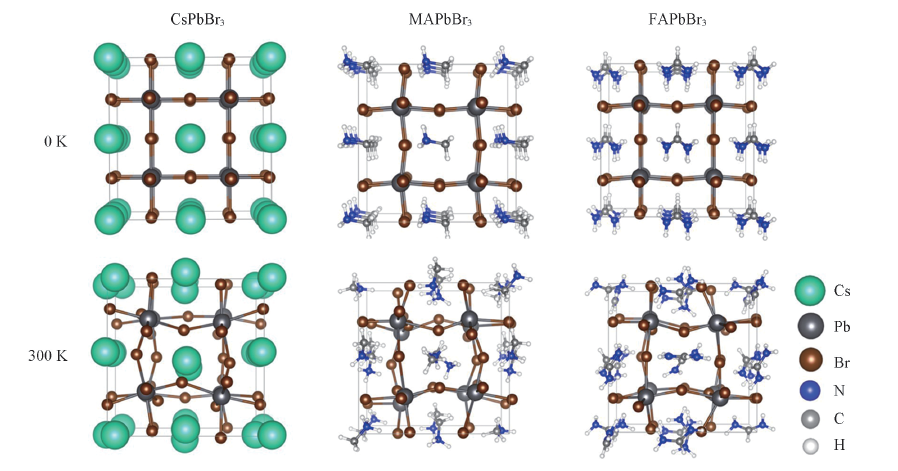
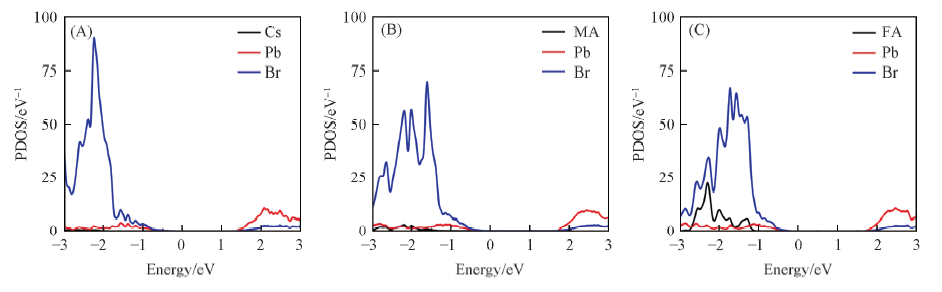
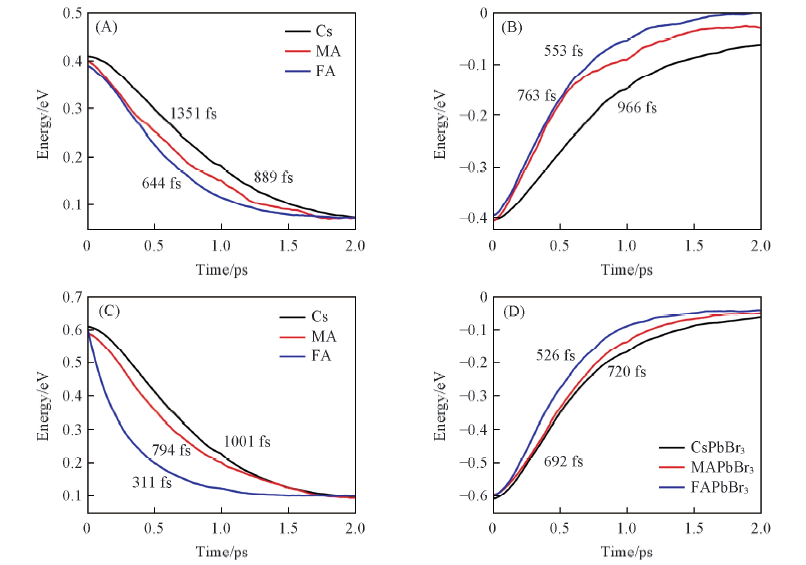
| [1] |
De Wolf S., Holovsky J., Moon S. J., Löper P., Niesen B., Ledinsky M., Haug F. J., Yum J. H., Ballif C ., J. Phys. Chem. Lett., 2014, 5, 1035— 1039
|
| [2] |
Shi D., Adinolfi V., Comin R., Yuan M., Alarousu E., Buin A., Chen Y., Hoogland S., Rothenberger A., Katsiev K ., Science, 2015, 347, 519— 522
|
| [3] |
Zhu H., Miyata K., Fu Y., Wang J., Joshi P. P., Niesner D., Williams K. W., Jin S., Zhu X. Y ., Science, 2016, 353, 1409— 1413
|
| [4] |
Zhu H., Fu Y., Meng F., Wu X., Gong Z., Ding Q., Gustafsson M. V., Trinh M. T., Jin S., Zhu X. Y ., Nature Materials, 2015, 14, 636— 642
|
| [5] |
Wang J., Dong J., Lu F., Sun C., Zhang Q., Wang N ., J. Mater. Chem. A, 2019, 7, 23563— 23576
|
| [6] |
Azpiroz J. M., Mosconi E., Bisquert J., de Angelis F ., Energy Environ. Sci., 2015, 8, 2118—2127
|
| [7] |
Dong X., Fang X., Lü M. H., Lin B. C., Zhang S., Wang Y., Yuan N. Y., Ding J. N ., Chinese Science Bulletin, 2016,1025
|
|
( 董旭, 房香, 吕明航, 林本才, 张帅, 王莹, 袁宁一, 丁建宁 . 科学通报, 2016,1025)
|
| [8] |
Chen C., Yang X. C., Liu W., ., CIESC J., 2017, 811— 820
|
|
( 陈超, 杨修春, 刘巍 . 化工学报, 2017, 811— 820)
|
| [9] |
Gan Y. S., Chen M. M., Wang Y. L., Wan L., Kong M. Q., Hu H., Wang S. M ., Materials Reports, 2018, 32, 4047—4050, 4078
|
|
( 甘一升, 陈苗苗, 王玉龙, 万丽, 孔梦琴, 胡航, 王世敏 . 材料导报, 2018, 32, 4047—4050, 4078)
|
| [10] |
Zhang D. F., Zheng L. L., Ma Y. Z., Wang S. F., Bian Z. Q., Huang C. H., Gong Q. H., Xiao L. X ., Acta Physica Sinica, 2015, 64, 118— 124
|
|
( 张丹霏, 郑灵灵, 马英壮, 王树峰, 卞祖强, 黄春辉, 龚旗煌, 肖立新 . 物理学报, 2015, 64, 118— 124)
|
| [11] |
Kojima A., Teshima K., Shirai Y., Miyasaka T ., J. Am. Chem. Soc., 2009, 131, 6050— 6051
|
| [12] |
NREL, Efficiency Chart., 2019,
|
| [13] |
Rühle S ., Sol. Energy, 2016, 130, 139— 147
|
| [14] |
Jeon N. J., Noh J. H., Yang W. S., Kim Y. C., Ryu S., Seo J., Seok S. I ., Nature, 2015, 517, 476— 480
|
| [15] |
Fu J., Xu Q., Han G., Wu B., Huan C. H. A., Leek M. L., Sum T. C ., Nat. Commun., 2017, 8, 1300
|
| [16] |
Chung H., Jung S. I., Kim H. J., Cha W., Sim E., Kim D., Koh W. K., Kim J ., Angew. Chem. Int. Ed. Engl., 2017, 56, 4160— 4164
|
| [17] |
Chen J., Messing M. E., Zheng K., Pullerits T ., J. Am. Chem. Soc., 2019, 141, 3532— 3540
|
| [18] |
Fang H. H., Adjokatse S., Shao S., Even J., Loi M. A ., Nat. Commun., 2018, 9, 243
|
| [19] |
Wang Y., Long R ., J. Phys. Chem. Lett., 2019, 10, 4354— 4361
|
| [20] |
Qiao L., Sun X., Long R ., J. Phys. Chem. Lett., 2019, 10, 672— 678
|
| [21] |
He J., Fang W. H., Long R., Prezhdo O. V ., ACS Energy Lett., 2018, 3,2070—2076
|
| [22] |
Hopper T. R., Gorodetsky A., Frost J. M., Müller C., Lovrincic R., Bakulin A. A ., ACS Energy Lett., 2018, 3, 2199— 2205
|
| [23] |
Petersilka M., Gossmann U., Gross E ., Phys. Rev. Lett., 1996, 76, 1212— 1215
|
| [24] |
Tully J. C ., J. Chem. Phys., 1990, 93, 1061— 1071
|
| [25] |
Craig C. F., Duncan W. R., Prezhdo O. V ., Phys. Rev. Lett., 2005, 95, 163001
|
| [26] |
Fischer S. A., Habenicht B. F., Madrid A. B., Duncan W. R., Prezhdo O. V ., J. Chem. Phys., 2011, 134, 024102
|
| [27] |
Kohn W., Sham L. J ., Phys. Rev., 1965, 140, A1133— A1138
|
| [28] |
Isborn C. M., Li X., Tully J. C ., J. Chem. Phys., 2007, 126, 134307
|
| [29] |
Hammes-Schiffer S., Tully J. C ., J. Chem. Phys., 1994, 101, 4657— 4667
|
| [30] |
Parandekar P. V., Tully J. C ., J. Chem. Phys., 2005, 122, 094102
|
| [31] |
Fabiano E., Keal T., Thiel W ., Chem. Phys., 2008, 349, 334— 347
|
| [32] |
Kapral R., Ciccotti G ., J. Chem. Phys., 1999, 110, 8919— 8929
|
| [33] |
Herman M. F., Kluk E ., Chem. Phys., 1984, 91, 27— 34
|
| [34] |
Tully J ., Faraday Discuss., 1998, 110, 407— 419
|
| [35] |
Akimov A. V., Prezhdo O. V ., J. Chem. Theory Comput., 2013, 9, 4959— 4972
|
| [36] |
Wang S., Luo Q., Fang W. H., Long R ., J. Phys. Chem. Lett., 2019, 10, 1234— 1241
|
| [37] |
Long R., Prezhdo O. V ., Nano Lett., 2014, 14, 3335— 3341
|
| [38] |
Li W., Sun Y. Y., Li L., Zhou Z., Tang J., Prezhdo O. V ., J. Am. Chem. Soc., 2018, 140, 15753— 15763
|
| [39] |
Long R., Prezhdo O. V ., Nano Lett., 2015, 15, 4274— 4281
|
| [40] |
Kresse G., Furthmüller J ., Phys. Rev. B, 1996, 54, 11169— 11186
|
| [41] |
Perdew J. P., Burke K., Ernzerhof M ., Phys. Rev. Lett., 1996, 77, 3865— 3868
|
| [42] |
Blöchl P. E ., Phys. Rev. B, 1994, 50, 17953— 17979
|
| [43] |
Monkhorst H. J., Pack J. D ., Phys. Rev. B, 1976, 13, 5188— 5192
|
| [44] |
Grimme S., Antony J., Ehrlich S., Krieg H ., J. Chem. Phys., 2010, 132, 154104
|
| [45] |
Rodová M., Brožek J., Knížek K., Nitsch K ., J. Therm. Anal. Calorim., 2003, 71, 667— 673
|
| [46] |
Elbaz G. A., Straus D. B., Semonin O. E., Hull T. D., Paley D. W., Kim P., Owen J. S., Kagan C. R., Roy X ., Nano Lett., 2017, 17, 1727— 1732
|
| [47] |
Zhumekenov A. A., Saidaminov M. I., Haque M. A., Alarousu E., Sarmah S. P., Murali B., Dursun I., Miao X. H., Abdelhady A. L., Wu T ., ACS Energy Lett., 2016, 1, 32— 37
|
| [48] |
Knop O. W., Wasylishen R. E., White M. A., Cameron T. S., Oort M. J. V ., Can. J. Chem., 1990, 68, 412— 422
|
| [49] |
Sun S., Isikgor F. H., Deng Z., Wei F., Kieslich G., Bristowe P. D., Ouyang J., Cheetham A. K ., ChemSusChem, 2017, 10, 3740— 3745
|
| [50] |
Jankowska J., Prezhdo O. V ., J. Phys. Chem. Lett., 2017, 8, 812— 818
|
| [51] |
Motta C., El-Mellouhi F., Sanvito S ., Phys. Rev. B, 2016, 93, 235412
|
| [52] |
El-Mellouhi F., Marzouk A., Bentria E. T., Rashkeev S. N., Kais S., Alharbi F. H ., ChemSusChem, 2016, 9, 2648— 2655
|
| [53] |
Ye Y., Run X., Xu H. T., Feng H., Fei X., Wang L. J ., Chin. Phys. B, 2015, 24, 116302— 116303
|
| [54] |
Tavernelli I., Tapavicza E., Rothlisberger U ., J. Chem. Phys., 2009, 130, 124107
|