

Chem. J. Chinese Universities ›› 2017, Vol. 38 ›› Issue (12): 2231.doi: 10.7503/cjcu20170363
• Physical Chemistry • Previous Articles Next Articles
LIU Ning1,2,3,*( ), ZHOU Cheng2, SHU Yuanjie2,3, WANG Bozhou2,3, WANG Wenliang1
), ZHOU Cheng2, SHU Yuanjie2,3, WANG Bozhou2,3, WANG Wenliang1
Received:2017-06-08
Online:2017-12-10
Published:2017-11-21
Contact:
LIU Ning
E-mail:flackliu@sina.com
Supported by:TrendMD:
LIU Ning, ZHOU Cheng, SHU Yuanjie, WANG Bozhou, WANG Wenliang. Molecular Dynamics Simulations on Crystal Morphology of N-Guanylurea-dinitramide†[J]. Chem. J. Chinese Universities, 2017, 38(12): 2231.
| Model | a/nm | b/nm | c/nm | α/(°) | β/(°) | γ/(°) | ρ/(g·cm-3) |
|---|---|---|---|---|---|---|---|
| Experimental[ | 1.367 | 0.933 | 0.614 | 90.00 | 90.00 | 90.00 | 1.775 |
| DREIDING | 1.400 | 0.919 | 0.615 | 90.00 | 90.00 | 90.00 | 1.755 |
| Relative error(%) | 2.41 | -1.50 | 0.01 | 0 | 0 | 0 | -1.13 |
Table 1 Comparison of the experimental and optimized lattice parameters of FOX-12
| Model | a/nm | b/nm | c/nm | α/(°) | β/(°) | γ/(°) | ρ/(g·cm-3) |
|---|---|---|---|---|---|---|---|
| Experimental[ | 1.367 | 0.933 | 0.614 | 90.00 | 90.00 | 90.00 | 1.775 |
| DREIDING | 1.400 | 0.919 | 0.615 | 90.00 | 90.00 | 90.00 | 1.755 |
| Relative error(%) | 2.41 | -1.50 | 0.01 | 0 | 0 | 0 | -1.13 |
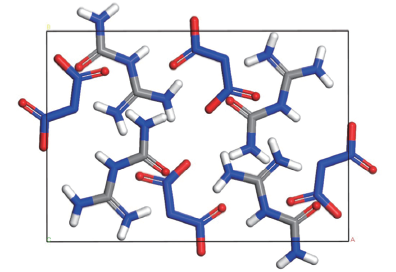
Fig.1 Unit cell structure of FOX-12
| (hkl) | Multiplicity | dhkl/nm | Eatt(total)/ (kJ·mol-1) | Eatt(vdW)/ (kJ·mol-1) | Eatt(H-bond)/ (kJ·mol-1) | D | Rhkl | Total facet area(%) |
|---|---|---|---|---|---|---|---|---|
| (110) | 4 | 0.771 | -196.11 | -78.44 | -117.67 | 46.85 | 1.22 | 33.95 |
| (200) | 2 | 0.683 | -161.32 | -100.46 | -60.86 | 38.54 | 1.00 | 18.27 |
| (201) | 2 | 0.457 | -189.20 | -128.46 | -60.74 | 45.20 | 1.17 | 25.40 |
| (011) | 2 | 0.513 | -221.85 | -121.14 | -100.71 | 53.00 | 1.38 | 14.70 |
| (002) | 1 | 0.307 | -205.82 | -205.53 | -0.29 | 49.17 | 1.28 | 6.42 |
| (111) | 4 | 0.480 | -237.76 | -119.84 | -117.92 | 56.80 | 1.47 | 1.26 |
Table 2 Crystal habit parameters of FOX-12 crystal in vacuum predicted by the AE model*
| (hkl) | Multiplicity | dhkl/nm | Eatt(total)/ (kJ·mol-1) | Eatt(vdW)/ (kJ·mol-1) | Eatt(H-bond)/ (kJ·mol-1) | D | Rhkl | Total facet area(%) |
|---|---|---|---|---|---|---|---|---|
| (110) | 4 | 0.771 | -196.11 | -78.44 | -117.67 | 46.85 | 1.22 | 33.95 |
| (200) | 2 | 0.683 | -161.32 | -100.46 | -60.86 | 38.54 | 1.00 | 18.27 |
| (201) | 2 | 0.457 | -189.20 | -128.46 | -60.74 | 45.20 | 1.17 | 25.40 |
| (011) | 2 | 0.513 | -221.85 | -121.14 | -100.71 | 53.00 | 1.38 | 14.70 |
| (002) | 1 | 0.307 | -205.82 | -205.53 | -0.29 | 49.17 | 1.28 | 6.42 |
| (111) | 4 | 0.480 | -237.76 | -119.84 | -117.92 | 56.80 | 1.47 | 1.26 |
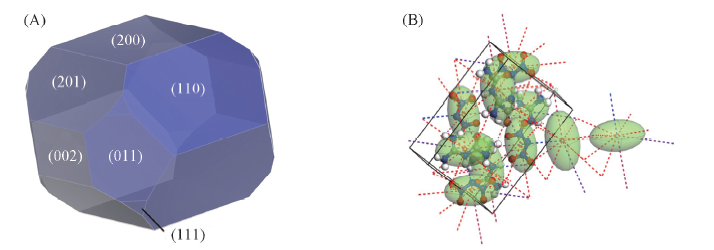
Fig.2 Crystal morphology(A) and crystal graph(B) of FOX-12 in vacuum predicted by the AE model
| Species | (hkl) | |||||
|---|---|---|---|---|---|---|
| (110) | (200) | (201) | (011) | (002) | (111) | |
| Aacc/nm2 | 0.5591 | 0.1485 | 0 | 0.2939 | 0 | 0 |
| Abox/nm2 | 11.1434 | 5.1552 | 10.0328 | 11.2639 | 7.6519 | 9.1605 |
Table 3 Accessible solvent surface area(Aacc) and total crystal face area(Abox) of FOX-12 surfaces calculated by the Connolly surface model
| Species | (hkl) | |||||
|---|---|---|---|---|---|---|
| (110) | (200) | (201) | (011) | (002) | (111) | |
| Aacc/nm2 | 0.5591 | 0.1485 | 0 | 0.2939 | 0 | 0 |
| Abox/nm2 | 11.1434 | 5.1552 | 10.0328 | 11.2639 | 7.6519 | 9.1605 |
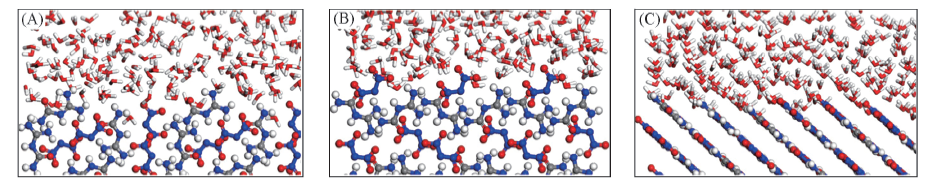
Fig.3 Configurations of FOX-12 surface-H2O interfaces from the MD equilibrium (A) (110); (B) (200); (C) (011).
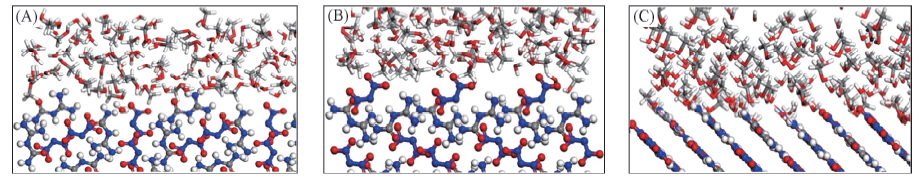
Fig.4 Configurations of FOX-12 surface-H2O/MeOH interfaces from the MD equilibrium (A) (110); (B) (200); (C) (011).
| Solvent | (hkl) | Etot/ (kJ·mol-1) | Esurf/ (kJ·mol-1) | Esolv/ (kJ·mol-1) | Eint/ (kJ·mol-1) | Es/ (kJ·mol-1) | (kJ·mol-1) | |
|---|---|---|---|---|---|---|---|---|
| H2O | (110) | -31114.1 | -29487.9 | 1862.1 | -3488.3 | -175.01 | -21.10 | 1.00 |
| (200) | -28123.8 | -26577.8 | 1630.5 | -3176.5 | -91.50 | -69.82 | 3.31 | |
| (011) | -31355.9 | -29334.1 | 2183.8 | -4205.6 | -109.75 | -112.10 | 5.31 | |
| H2O/MeOH | (110) | -27888.6 | -29487.9 | 5890.0 | -4290.6 | -215.28 | 19.17 | |
| (200) | -24927.5 | -26577.8 | 5657.9 | -4007.7 | -115.45 | -45.88 | 1.00 | |
| (011) | -28079.2 | -29334.1 | 6057.7 | -4802.8 | -125.32 | -96.53 | 2.10 |
Table 4 Interaction energies, modified attachment energies and relative growth rates of FOX-12 crystal habit faces in solvents
| Solvent | (hkl) | Etot/ (kJ·mol-1) | Esurf/ (kJ·mol-1) | Esolv/ (kJ·mol-1) | Eint/ (kJ·mol-1) | Es/ (kJ·mol-1) | (kJ·mol-1) | |
|---|---|---|---|---|---|---|---|---|
| H2O | (110) | -31114.1 | -29487.9 | 1862.1 | -3488.3 | -175.01 | -21.10 | 1.00 |
| (200) | -28123.8 | -26577.8 | 1630.5 | -3176.5 | -91.50 | -69.82 | 3.31 | |
| (011) | -31355.9 | -29334.1 | 2183.8 | -4205.6 | -109.75 | -112.10 | 5.31 | |
| H2O/MeOH | (110) | -27888.6 | -29487.9 | 5890.0 | -4290.6 | -215.28 | 19.17 | |
| (200) | -24927.5 | -26577.8 | 5657.9 | -4007.7 | -115.45 | -45.88 | 1.00 | |
| (011) | -28079.2 | -29334.1 | 6057.7 | -4802.8 | -125.32 | -96.53 | 2.10 |
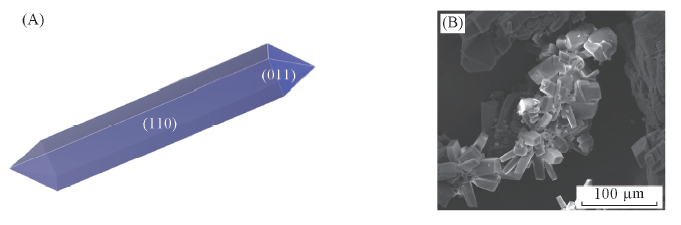
Fig.5 Predicted crystal morphology(A) and SEM image(B) of FOX-12 from H2O
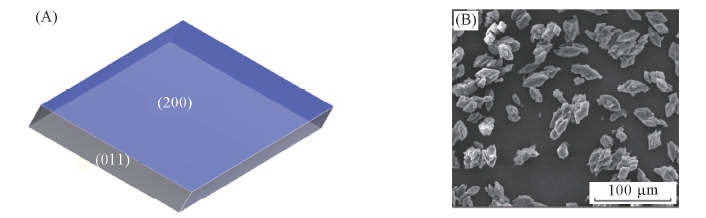
Fig.6 Predicted crystal morphology(A) and SEM image(B) of FOX-12 from H2O/MeOH
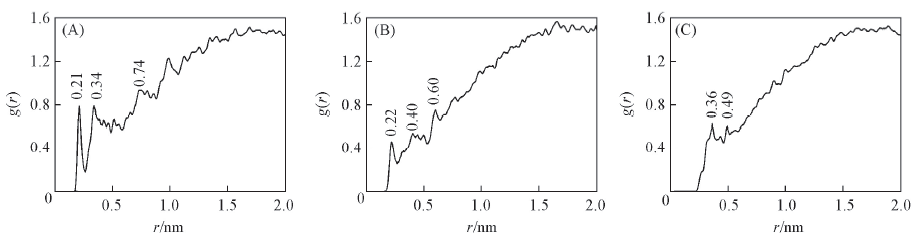
Fig.7 RDF analysis of H2O molecules and FOX-12 crystal for the (110) face (A) HFOX-12…OH2O; (B) HH2O…O FOX-12; (C) HH2O…NFOX-12.
| [1] | Ghee A.H., Santhosh G., Advances in Energetic Dinitramides—An Emerging Class of Inorganic Oxidizers, World Scientific Press, Singapore, 2008 |
| [2] | Östmark H., Bemm U., Bergman H., Langlet A., Thermochim. Acta, 2002, 384, 253—259 |
| [3] | Wang K., Chen J. G., Wang B. Z., Lv J., Wang W. L., Liu F. Y., Zhou C., Lian P., Liu Z. W., Liu Z. T., Chem. J. Chinese Universities, 2015, 36(3), 531—538 |
| (王宽, 陈建刚, 王伯周, 吕剑, 王文亮, 刘峰毅, 周诚, 廉鹏, 刘忠文, 刘昭铁. 高等学校化学学报, 2015, 36(3), 531—538) | |
| [4] | Kempa P. B., Herrmann M., Fuhr I., 40th International Annual Conference of ICT, Karlsruhe,2009, 40-1—40-11 |
| [5] | Zhang J. Q., Zhao W. W., Ji T. Z., Gao H. X., Hu R. Z., J. Therm. Anal. Calorim., 2014, 115, 641—646 |
| [6] | Li N., Zhao F. Q., Luo Y., Gao H. X., Xiao L. B., Hu R. Z., Ju R. H., J. Therm. Anal. Calorim., 2014, 115, 869—873 |
| [7] | Wang B. Z., Liu Q., Zhang Z. Z., Ji Y. P., Zhu C. H., Chin. J. Energ. Mater., 2004, 12(1), 38—39 |
| (王伯周, 刘愆, 张志忠, 姬月萍, 朱春华. 含能材料, 2004, 12(1), 38—39) | |
| [8] | Santhosh G., Soumyamol P. B., Sreejitha M., Reshmi S., Thermochim. Acta, 2016, 632, 46—51 |
| [9] | Perreault N. N., Halasz A., Thiboutot S., Ampleman G., Hawari J., Environ. Sci. Technol., 2013, 47, 5193—5198 |
| [10] | Lei Y. P., Yang S. Q., Xu S. L., Zhang T., Chin. J. Energ. Mater., 2007, 15(3), 289—293 |
| (雷永鹏, 阳世清, 徐松林, 张彤. 含能材料, 2007, 15(3), 289—293) | |
| [11] | Duan X., Wei C., Liu Y., Pei C., J. Hazard. Mater., 2010, 174, 175—180 |
| [12] | Yan T., Wang J. H., Liu Y. C., Zhao J., Yuan J. M., Guo J. H., J. Cryst. Growth, 2015, 430, 7—13 |
| [13] | Chen G., Xia M., Lei W., Wang F., Gong X., J. Phys. Chem. A, 2014, 118, 11471—11478 |
| [14] | Chen G., Chen C., Xia M., Lei W., Wang F., Gong X., RSC Adv., 2015, 5, 25581—25589 |
| [15] | Zhang C., Ji C., Li H., Zhou Y., Xu J., Xu R., Li J., Luo Y., Cryst. Growth Des., 2013, 13, 282—290 |
| [16] | Shim H. M., Koo K. K., Cryst. Growth Des., 2014, 14, 1802—1810 |
| [17] | Liu N., Li Y., Zeman S., Shu Y., Wang B., Zhou Y., Zhao Q., Wang W., Cryst. Eng. Comm., 2016, 18, 2843—2851 |
| [18] | Zhao Q., Liu N., Wang B., Wang W., RSC Adv., 2016, 6, 59141—59149 |
| [19] | Liu N., Wang B. Z., Shu Y. J., Wu Z. K., Zhou Q., Zhao Q. L., Wang W. L., Chinese Journal of Explosives & Propellants,2016, 39(2), 40—44 |
| (刘宁, 王伯周, 舒远杰, 武宗凯, 周群, 赵强莉, 王文亮. 火炸药学报, 2016, 39(2), 40—44) | |
| [20] | Stephen L. M., Barry D. O., William A. G. III, J. Phys. Chem., 1990, 94, 8897—8909 |
| [21] | Material Studio 5.5, Acceryls Inc., San Diego, 2010 |
| [22] | Andersen H. C., J. Chem. Phys., 1980, 72, 2384—2393 |
| [23] | Hartman P., Bennema P., J. Cryst. Growth, 1980, 49, 145—156 |
| [24] | Hartman P., J. Cryst. Growth, 1980, 49, 166—170 |
| [25] | Berkovitch-Yellin Z., J. Am. Chem. Soc., 1985, 107, 8239—8253 |
| [26] | Connolly M. L., Science, 1983, 221, 709—713 |
| [1] | GAO Zhiwei, LI Junwei, SHI Sai, FU Qiang, JIA Junru, AN Hailong. Analysis of Gating Characteristics of TRPM8 Channel Based on Molecular Dynamics [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220080. |
| [2] | HU Bo, ZHU Haochen. Dielectric Constant of Confined Water in a Bilayer Graphene Oxide Nanosystem [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210614. |
| [3] | ZHANG Mi, TIAN Yafeng, GAO Keli, HOU Hua, WANG Baoshan. Molecular Dynamics Simulation of the Physicochemical Properties of Trifluoromethanesulfonyl Fluoride Dielectrics [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220424. |
| [4] | LEI Xiaotong, JIN Yiqing, MENG Xuanyu. Prediction of the Binding Site of PIP2 in the TREK-1 Channel Based on Molecular Modeling [J]. Chem. J. Chinese Universities, 2021, 42(8): 2550. |
| [5] | LI Congcong, LIU Minghao, HAN Jiarui, ZHU Jingxuan, HAN Weiwei, LI Wannan. Theoretical Study of the Catalytic Activity of VmoLac Non-specific Substrates Based on Molecular Dynamics Simulations [J]. Chem. J. Chinese Universities, 2021, 42(8): 2518. |
| [6] | ZENG Yonghui, YAN Tianying. Vibrational Density of States Analysis of Proton Hydration Structure [J]. Chem. J. Chinese Universities, 2021, 42(6): 1855. |
| [7] | QI Renrui, LI Minghao, CHANG Hao, FU Xueqi, GAO Bo, HAN Weiwei, HAN Lu, LI Wannan. Theoretical Study on the Unbinding Pathway of Xanthine Oxidase Inhibitors Based on Steered Molecular Dynamics Simulation [J]. Chem. J. Chinese Universities, 2021, 42(3): 758. |
| [8] | LIU Aiqing, XU Wensheng, XU Xiaolei, CHEN Jizhong, AN Lijia. Molecular Dynamics Simulation of Polymer/rod Nanocomposite [J]. Chem. J. Chinese Universities, 2021, 42(3): 875. |
| [9] | QU Siying, XU Qin. Different Roles of Some Key Residues in the S4 Pocket of Coagulation Factor Xa for Rivaroxaban Binding † [J]. Chem. J. Chinese Universities, 2019, 40(9): 1918. |
| [10] | MA Yucong, FAN Baomin, WANG Manman, YANG Biao, HAO Hua, SUN Hui, ZHANG Huijuan. Two-step Preparation of Trazodone and Its Corrosion Inhibition Mechanism for Carbon Steel [J]. Chem. J. Chinese Universities, 2019, 40(8): 1706. |
| [11] | ZHANG Zhang,WANG Dong,WANG Xiaolei,XU Yan. Regulation of Ester Synthesis Activity of Rhizopus chinensis Lipase† [J]. Chem. J. Chinese Universities, 2019, 40(4): 747. |
| [12] | MA Lan,RONG Jingjing,ZHU Youliang,HUANG Yineng,SUN Zhaoyan. Simulation on the Dynamic Process of Formation of Particle Cluster by Generalized Exponential Model† [J]. Chem. J. Chinese Universities, 2019, 40(1): 195. |
| [13] | ZHU Jingxuan,YU Zhengfei,LIU Ye,ZHAN Dongling,HAN Jiarui,TIAN Xiaopian,HAN Weiwei. Exploration of Increasing the Non-specificity Substrates Activity for the Phosphotriesterase-like Lactonase Using Molecular Dynamics Simulations† [J]. Chem. J. Chinese Universities, 2019, 40(1): 138. |
| [14] | WU Hongmei,LI Huiting,LI Yongcheng,WANG Hongqing,WANG Meng. Using Group Contribution Method and Molecular Dynamics to Predict the Glass Transition Temperature of Poly(p-phenylene isophthalamide)† [J]. Chem. J. Chinese Universities, 2019, 40(1): 180. |
| [15] | LIU Yanfang, YANG Hua, ZHANG Hui. Molecular Dynamics Simulation on the Orientation of Alkane Mixture on Graphene† [J]. Chem. J. Chinese Universities, 2018, 39(8): 1729. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||