高等学校化学学报
2026, 47 (
):
20250299-.
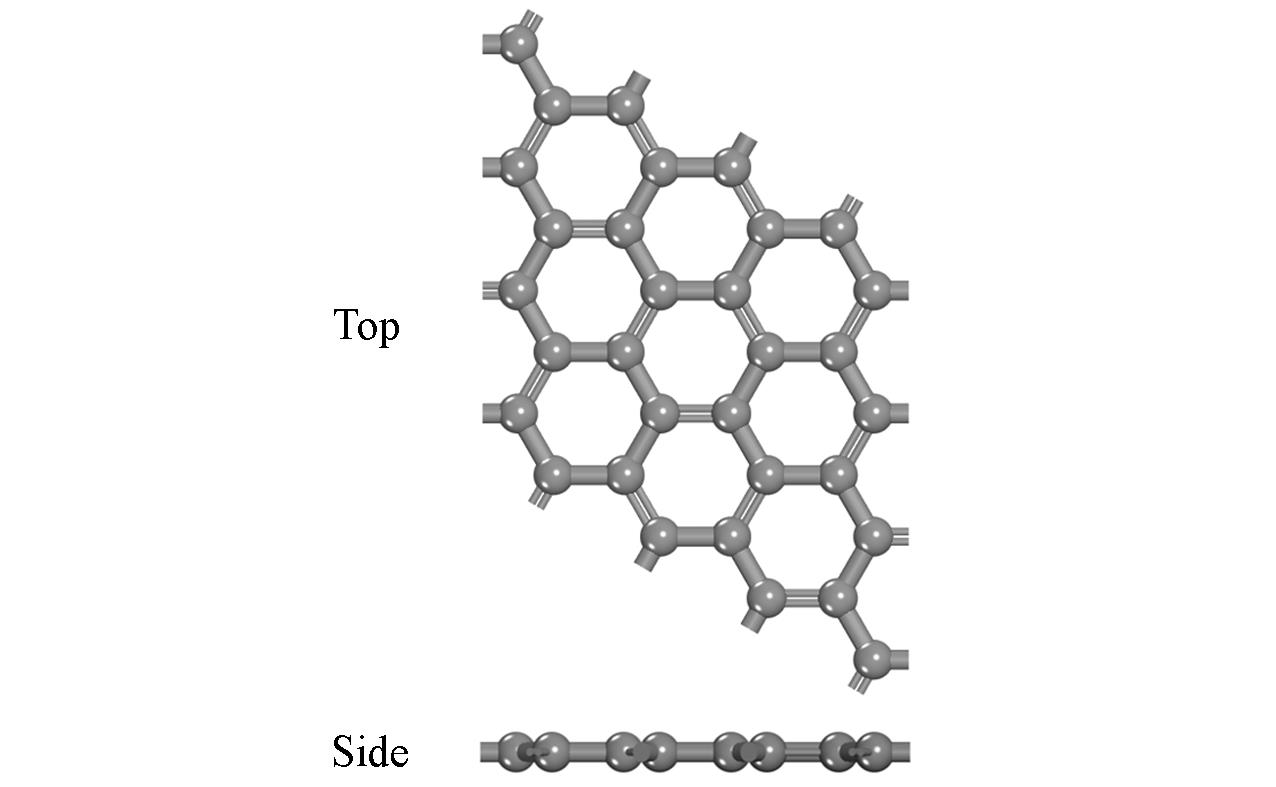
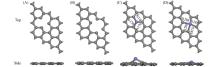
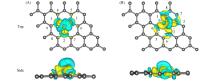
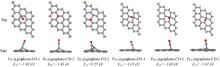
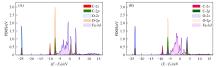
基于密度泛函理论(DFT), 研究了缺陷石墨烯限域调控Fe原子的结构、 电子性质、 CO吸附和活化性能, 揭示了Fe原子不同配位环境对费托活化性能的影响. 结果显示, Fe掺杂单原子缺陷石墨烯(FeC@graphene)和Fe掺杂二原子缺陷石墨烯(Fe2C@graphene)的结合能分别为-7.49和-6.50 eV, 表明FeC@graphene的结构更稳定. 由于FeC@graphene的态密度(DOS)向左偏移值大于Fe2C@graphene(1.5 eV> 0.8 eV), FeC@graphene结构的能量更低, 所以结构更稳定. CO在FeC@graphene和Fe2C@graphene的吸附能分别为-1.43和-1.69 eV, 表明CO更稳定地吸附在Fe2C@graphene上. FeC@graphene和Fe2C@graphene的d带中心值分别为-1.26和-0.83 eV; 能带带隙分别为0.45和0.01 eV, d带中心越接近费米能级, 带隙越小, 越有利于物种吸附, 所以CO更容易吸附在Fe2C@graphene上. Fe2C@graphene-CO带隙增加0.25 eV, 而FeC@graphene-CO带隙降低0.04 eV; FeC@graphene-CO和Fe2C@graphene-CO的集成晶体轨道哈密顿布居(ICOHP)值分别为-1.99和-2.50 eV, 表明Fe2C@graphene与CO之间的相互作用更强, 而强相互作用不利于CO活化. 在FeC@graphene和Fe2C@graphene结构中, CO活化的最佳路径为CO* → CHO* → CH* + O*, 有效能垒分别为2.53和3.50 eV, CO在FeC@graphene上更容易活化. 因此, 活性中心Fe原子的三配位结构更稳定且有利于提高费托活性.







{kind=link}