

高等学校化学学报 ›› 2024, Vol. 45 ›› Issue (4): 20240036.doi: 10.7503/cjcu20240036
• 物理化学 • 上一篇
聂建航1, 王天奇1, 金丽1, 张建坡1( ), 张红星2, 白福全2
), 张红星2, 白福全2
收稿日期:2024-01-22
出版日期:2024-04-10
发布日期:2024-03-05
通讯作者:
张建坡
E-mail:zhangjp725@126.com
基金资助:
NIE Jianhang1, WANG Tianqi1, JIN Li1, ZHANG Jianpo1(), ZHANG Hongxing2, BAI Fuquan2
Received:2024-01-22
Online:2024-04-10
Published:2024-03-05
Contact:
ZHANG Jianpo
E-mail:zhangjp725@126.com
Supported by:摘要:
对一类环金属Ir(III) [(C^N)2Ir(A^A)]配合物[C^N=ptaz(1, 2, 4), mhtz(3), ptaz=3,4,5-三苯基-4H-1,2,4-三唑, mhtz=1,3-双甲基-5-苯基-1H-1,2,4-三唑; A^A=pzpy(1), npzpy(2, 3), bicb(4), pzpy=2-(1H-吡唑-1-基)吡啶, npzpy=4-二甲基氨基-2-(1H-吡唑-1-基)吡啶, bicb=3,3′-亚甲基双(1-甲基-1H-咪唑-2-亚基)]的结构、 光谱特征和磷光量子效率进行了理论研究. 计算方法探究表明, 基于B3LYP泛函优化的基态结构和单激发组态相互作用(CIS)方法得到的激发态结构计算的吸收和发射光谱更准确. 配合物1~4的最低吸收峰和发射峰分别位于408, 376, 382, 365 nm和503, 506, 468, 511 nm处, 其HOMOs主要由金属和C^N配体占据, 而配合物1~3的LUMOs由A^A配体的π反键轨道组成, 配合物4的LUMO存在于C^N配体上. 因此, 配合物4的最低吸收峰和发射峰具有与配合物1~3不同的金属到配体和配体内部(MLCT/ILCT)的混合跃迁性质, 非共轭N^N配体的引入显著消弱了其在 跃迁过程中的贡献程度. 配合物1~4的量子效率取决于非辐射跃迁速率常数knr, 这与它们重组能的贡献 [4569 cm‒1(3)>2583 cm‒1(1)>1232 cm‒1(2)>975 cm‒1(4)]相一致, 表明主配体的体积和辅助配体的共轭能力都能影响配合物的磷光量子效率.
中图分类号:
TrendMD:
聂建航, 王天奇, 金丽, 张建坡, 张红星, 白福全. 环金属配体Ir(III)配合物结构、 光谱和量子效率的理论研究. 高等学校化学学报, 2024, 45(4): 20240036.
NIE Jianhang, WANG Tianqi, JIN Li, ZHANG Jianpo, ZHANG Hongxing, BAI Fuquan. Theoretical Study of Structure, Spectra and Quantum Efficiency for a Series of Iridium(III) Complexes with the Cyclometalating Ligand. Chem. J. Chinese Universities, 2024, 45(4): 20240036.
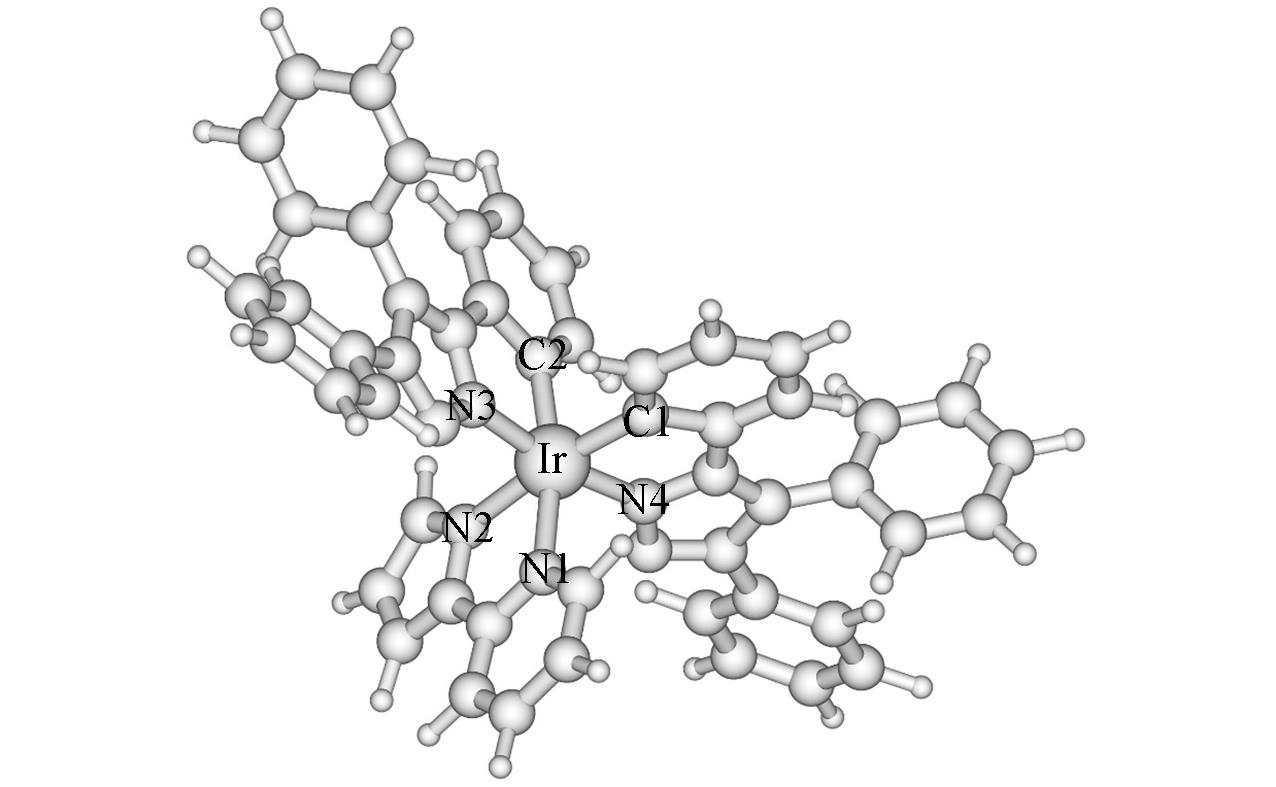
Fig.1 Optimized geometry of complex 1 in the ground state
| Species | B3LYP | CAM⁃B3LYP | M062X | Expt.[ | |||
|---|---|---|---|---|---|---|---|
| Cal | δ | Cal | δ | Cal | δ | ||
| d(Ir—C1)/nm | 0.2046 | 1.7 | 0.2039 | 1.4 | 0.2016 | 0.2 | 0.2011 |
| d(Ir—C2)/nm | 0.2047 | 1.8 | 0.2039 | 1.4 | 0.2015 | 0.2 | 0.2010 |
| d(Ir—N1)/nm | 0.2136 | 2.9 | 0.2164 | 4.3 | 0.2196 | 5.9 | 0.2074 |
| d(Ir—N2)/nm | 0.2091 | 0.1 | 0.2123 | 1.4 | 0.2160 | 3.2 | 0.2093 |
| d(Ir—N4)/nm | 0.2043 | 1.6 | 0.2033 | 1.1 | 0.2037 | 1.3 | 0.2010 |
| ∠N3—Ir—C2/(°) | 79.0 | 1.8 | 79.1 | 1.9 | 79.6 | 2.6 | 77.6 |
| ∠N1—Ir—N2/(°) | 75.6 | 1.7 | 75.2 | 2.2 | 74.9 | 2.6 | 76.9 |
Table 1 Structure parameters of complex 1 in the ground state by different functionals, the percent error( δ ) and experimental data
| Species | B3LYP | CAM⁃B3LYP | M062X | Expt.[ | |||
|---|---|---|---|---|---|---|---|
| Cal | δ | Cal | δ | Cal | δ | ||
| d(Ir—C1)/nm | 0.2046 | 1.7 | 0.2039 | 1.4 | 0.2016 | 0.2 | 0.2011 |
| d(Ir—C2)/nm | 0.2047 | 1.8 | 0.2039 | 1.4 | 0.2015 | 0.2 | 0.2010 |
| d(Ir—N1)/nm | 0.2136 | 2.9 | 0.2164 | 4.3 | 0.2196 | 5.9 | 0.2074 |
| d(Ir—N2)/nm | 0.2091 | 0.1 | 0.2123 | 1.4 | 0.2160 | 3.2 | 0.2093 |
| d(Ir—N4)/nm | 0.2043 | 1.6 | 0.2033 | 1.1 | 0.2037 | 1.3 | 0.2010 |
| ∠N3—Ir—C2/(°) | 79.0 | 1.8 | 79.1 | 1.9 | 79.6 | 2.6 | 77.6 |
| ∠N1—Ir—N2/(°) | 75.6 | 1.7 | 75.2 | 2.2 | 74.9 | 2.6 | 76.9 |
| Species | B3LYP | CAM⁃B3LYP | M062X | CIS | Expt. [ |
|---|---|---|---|---|---|
| Absorption/nm | 408 | 306 | 301 | — | 384 |
| Emission/nm | 571 | 412 | 518 | 503 | 486 |
Table 2 Calculated lowest absorption and emission wavelengths of complex 1 in CH3CN media by different method
| Species | B3LYP | CAM⁃B3LYP | M062X | CIS | Expt. [ |
|---|---|---|---|---|---|
| Absorption/nm | 408 | 306 | 301 | — | 384 |
| Emission/nm | 571 | 412 | 518 | 503 | 486 |
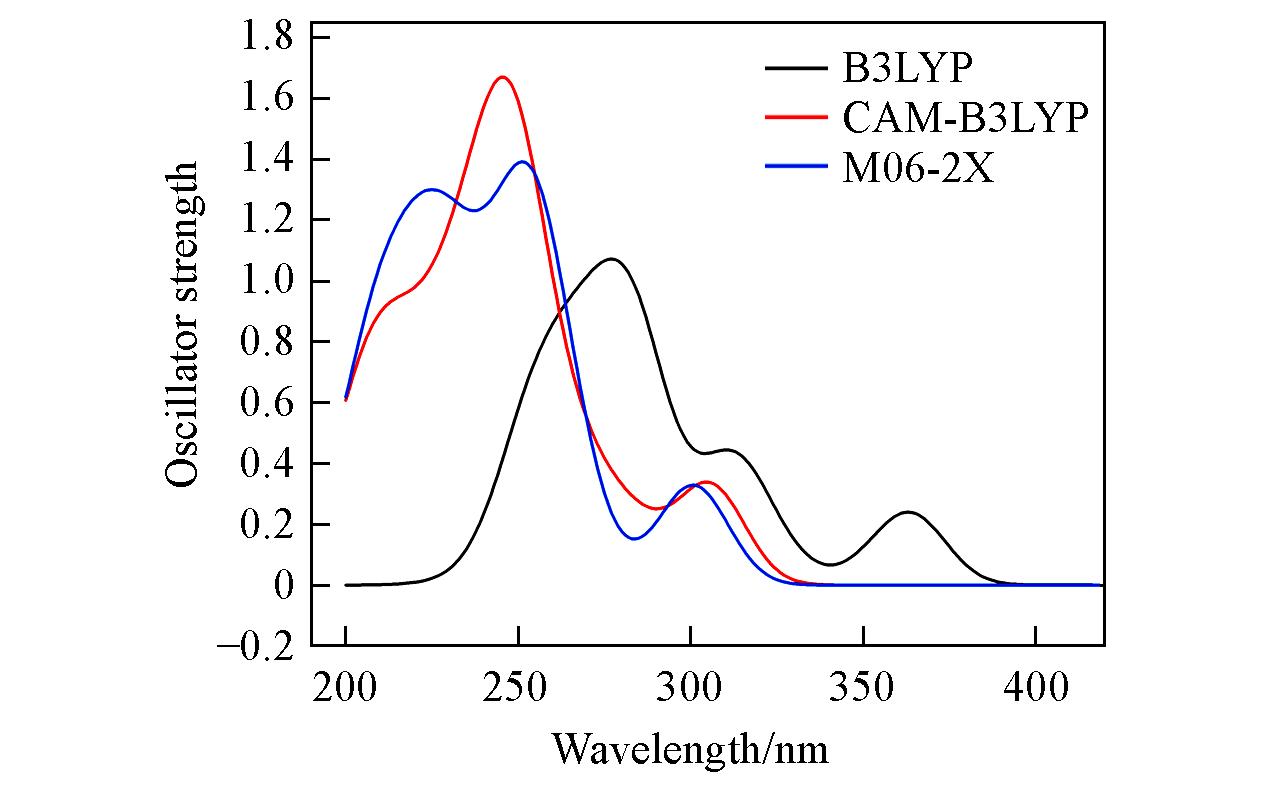
Fig.2 Simulated absorption spectra of complex 1 in CH3CN media using different functionals
| Species | 1 | Expt.[ | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| S0 | T1 | S0 | T1 | S0 | T1 | S0 | T1 | ||
| d(Ir—C1)/nm | 0.2047 | 0.2045 | 0.2011 | 0.2045 | 0.2026 | 0.2044 | 0.2041 | 0.2091 | 0.2088 |
| d(Ir—C2)/nm | 0.2048 | 0.2026 | 0.2010 | 0.2048 | 0.2047 | 0.2045 | 0.2040 | 0.2100 | 0.2094 |
| d(Ir—C3)/nm | 0.2127 | 0.2172 | |||||||
| d(Ir—C4)/nm | 0.2123 | 0.2160 | |||||||
| d(Ir—N1)/nm | 0.2136 | 0.2003 | 0.2074 | 0.2184 | 0.2121 | 0.2198 | 0.2103 | ||
| d(Ir—N2)/nm | 0.2091 | 0.2052 | 0.2093 | 0.2141 | 0.2135 | 0.2155 | 0.2151 | ||
| d(Ir—N3)/nm | 0.2041 | 0.2209 | 0.1973 | 0.2043 | 0.2081 | 0.2073 | 0.2092 | 0.2051 | 0.2071 |
| d(Ir—N4)/nm | 0.2043 | 0.2160 | 0.2010 | 0.2042 | 0.2054 | 0.2076 | 0.2079 | 0.2.060 | 0.2082 |
| ∠N3—Ir—C2)/(°) | 79.0 | 80.6 | 77.6 | 79.0 | 80.7 | 79.6 | 79.7 | 78.1 | 77.7 |
| ∠N4—Ir—C1)/(°) | 79.0 | 79.0 | 81.5 | 79.0 | 79.0 | 79.6 | 79.7 | 77.9 | 77.6 |
| ∠N1—Ir—N2)/(°) | 75.6 | 75.1 | 76.9 | 75.4 | 74.9 | 75.0 | 76.8 | ||
| ∠C3—Ir—C4)/(°) | 84.4 | 84.1 | |||||||
| ∠C1—Ir—C2—N3/(°) | 95.5 | 95.0 | 94.8 | 94.8 | 94.6 | 95.7 | 93.9 | 94.7 | |
| ∠N4—Ir—N1—N2/(°) | 95.0 | 94.8 | 94.8 | 94.5 | 98.8 | 99.8 | |||
| ∠C1—Ir—C3—C4/(°) | 97.8 | 97.8 | |||||||
Table 3 Main optimized geometry structural parameters of complexes 1—4 in the S0 and T1 states by B3LYP and CIS methods, respectively
| Species | 1 | Expt.[ | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| S0 | T1 | S0 | T1 | S0 | T1 | S0 | T1 | ||
| d(Ir—C1)/nm | 0.2047 | 0.2045 | 0.2011 | 0.2045 | 0.2026 | 0.2044 | 0.2041 | 0.2091 | 0.2088 |
| d(Ir—C2)/nm | 0.2048 | 0.2026 | 0.2010 | 0.2048 | 0.2047 | 0.2045 | 0.2040 | 0.2100 | 0.2094 |
| d(Ir—C3)/nm | 0.2127 | 0.2172 | |||||||
| d(Ir—C4)/nm | 0.2123 | 0.2160 | |||||||
| d(Ir—N1)/nm | 0.2136 | 0.2003 | 0.2074 | 0.2184 | 0.2121 | 0.2198 | 0.2103 | ||
| d(Ir—N2)/nm | 0.2091 | 0.2052 | 0.2093 | 0.2141 | 0.2135 | 0.2155 | 0.2151 | ||
| d(Ir—N3)/nm | 0.2041 | 0.2209 | 0.1973 | 0.2043 | 0.2081 | 0.2073 | 0.2092 | 0.2051 | 0.2071 |
| d(Ir—N4)/nm | 0.2043 | 0.2160 | 0.2010 | 0.2042 | 0.2054 | 0.2076 | 0.2079 | 0.2.060 | 0.2082 |
| ∠N3—Ir—C2)/(°) | 79.0 | 80.6 | 77.6 | 79.0 | 80.7 | 79.6 | 79.7 | 78.1 | 77.7 |
| ∠N4—Ir—C1)/(°) | 79.0 | 79.0 | 81.5 | 79.0 | 79.0 | 79.6 | 79.7 | 77.9 | 77.6 |
| ∠N1—Ir—N2)/(°) | 75.6 | 75.1 | 76.9 | 75.4 | 74.9 | 75.0 | 76.8 | ||
| ∠C3—Ir—C4)/(°) | 84.4 | 84.1 | |||||||
| ∠C1—Ir—C2—N3/(°) | 95.5 | 95.0 | 94.8 | 94.8 | 94.6 | 95.7 | 93.9 | 94.7 | |
| ∠N4—Ir—N1—N2/(°) | 95.0 | 94.8 | 94.8 | 94.5 | 98.8 | 99.8 | |||
| ∠C1—Ir—C3—C4/(°) | 97.8 | 97.8 | |||||||
| Complex | Transition | Config(CIcoeff) | λmin/nm(Em/eV) | Oscillator | Assignment | Expt. |
|---|---|---|---|---|---|---|
| 1 | X1A→A1A | 201→202(0.70) | 408(3.04) | 0.0008 | MLCT/LLCT | 384[ |
| X1A→B1A | 201→203(0.66) | 365(3.40) | 0.1835 | MLCT/LLCT | 359[ | |
| X1A→C1A | 200→204(0.54) | 315(3.94) | 0.1387 | MLCT/ILCT | ||
| X1A→D1A | 196→202(0.47) | 285(4.35) | 0.2437 | LLCT | ||
| 2 | X1A→A1A | 213→214(0.70) | 376(3.31) | 0.0272 | MLCT/LLCT | 388[ |
| X1A→B1A | 213→215(0.67) | 367(3.38) | 0.1560 | MLCT/ILCT | 363[ | |
| X1A→C1A | 210→216(0.38) | 288(4.30) | 0.4026 | MLCT/LLCT | ||
| X1A→D1A | 206→214(0.54) | 249(4.99) | 0.5938 | LLCT | ||
| 3 | X1A→A1A | 149→150(0.70) | 382(3.24) | 0.0007 | MLCT/LLCT | 380[ |
| X1A→B1A | 148→150(0.58) | 345(3.59) | 0.0761 | MLCT/LLCT | 355[ | |
| X1A→C1A | 148→153(0.55) | 278(4.45) | 0.1812 | MLCT/LLCT | ||
| X1A→D1A | 142→150(0.55) | 251(4.93) | 0.1385 | LLCT | ||
| 4 | X1A→A1A | 210→211(0.68) | 365(3.40) | 0.1507 | MLCT/ILCT | 355[ |
| X1A→B1A | 209→212(0.49) | 320(3.87) | 0.1661 | MLCT/ILCT | ||
| X1A→C1A | 206→211(0.59) | 280(4.44) | 0.1882 | MLCT/LLCT | ||
| X1A→D1A | 203→211(0.44) | 258(4.81) | 0.1342 | LLCT |
Table 4 Calculated absorptions of complexes 1—4 in CH3CN media by B3LYP and CIS methods and experimental values
| Complex | Transition | Config(CIcoeff) | λmin/nm(Em/eV) | Oscillator | Assignment | Expt. |
|---|---|---|---|---|---|---|
| 1 | X1A→A1A | 201→202(0.70) | 408(3.04) | 0.0008 | MLCT/LLCT | 384[ |
| X1A→B1A | 201→203(0.66) | 365(3.40) | 0.1835 | MLCT/LLCT | 359[ | |
| X1A→C1A | 200→204(0.54) | 315(3.94) | 0.1387 | MLCT/ILCT | ||
| X1A→D1A | 196→202(0.47) | 285(4.35) | 0.2437 | LLCT | ||
| 2 | X1A→A1A | 213→214(0.70) | 376(3.31) | 0.0272 | MLCT/LLCT | 388[ |
| X1A→B1A | 213→215(0.67) | 367(3.38) | 0.1560 | MLCT/ILCT | 363[ | |
| X1A→C1A | 210→216(0.38) | 288(4.30) | 0.4026 | MLCT/LLCT | ||
| X1A→D1A | 206→214(0.54) | 249(4.99) | 0.5938 | LLCT | ||
| 3 | X1A→A1A | 149→150(0.70) | 382(3.24) | 0.0007 | MLCT/LLCT | 380[ |
| X1A→B1A | 148→150(0.58) | 345(3.59) | 0.0761 | MLCT/LLCT | 355[ | |
| X1A→C1A | 148→153(0.55) | 278(4.45) | 0.1812 | MLCT/LLCT | ||
| X1A→D1A | 142→150(0.55) | 251(4.93) | 0.1385 | LLCT | ||
| 4 | X1A→A1A | 210→211(0.68) | 365(3.40) | 0.1507 | MLCT/ILCT | 355[ |
| X1A→B1A | 209→212(0.49) | 320(3.87) | 0.1661 | MLCT/ILCT | ||
| X1A→C1A | 206→211(0.59) | 280(4.44) | 0.1882 | MLCT/LLCT | ||
| X1A→D1A | 203→211(0.44) | 258(4.81) | 0.1342 | LLCT |
| Complex | Orbital | Energy/eV | Composition(%) | Main bond type | ||
|---|---|---|---|---|---|---|
| Ir | C^N | A^A | ||||
| 1 | 204 | -3.390 | 1.7 | 93.9 | 4.4 | π*(ptaz) |
| 203 | -3.597 | 1.1 | 2.8 | 96.1 | π*(pzpy) | |
| 202(L) | -4.080 | 3.9 | 2.5 | 93.6 | π*(pzpy) | |
| 201(H) | -7.516 | 42.0 | 54.3 | 3.6 | d(Ir)+π(ptaz) | |
| 200 | -7.777 | 28.6 | 67.3 | 4.2 | d(Ir)+π(ptaz) | |
| 196 | -8.619 | 4.1 | 51.5 | 44.4 | π(ptaz)+π(pzpy) | |
| 2 | 216 | -3.180 | 1.6 | 97.4 | 0.9 | π*(ptaz) |
| 215 | -3.288 | 1.4 | 95.4 | 3.2 | π*(ptaz) | |
| 214(L) | -3.625 | 2.2 | 2.6 | 95.1 | π*(npzpy) | |
| 213(H) | -7.279 | 44.1 | 52.2 | 3.7 | d(Ir)+π(ptaz) | |
| 210 | -8.083 | 19.4 | 53.0 | 27.6 | d(Ir)+π(ptaz)+π(npzpy) | |
| 206 | -8.754 | 0.8 | 98.4 | 0.8 | π(ptaz) | |
| 3 | 153 | -3.339 | 3.2 | 88.5 | 8.3 | π*(mhtz) |
| 150(L) | -4.296 | 3.0 | 2.2 | 94.9 | π*(npzpy) | |
| 149(H) | -7.762 | 43.4 | 48.9 | 7.7 | d(Ir)+π(mhtz) | |
| 148 | -8.149 | 46.7 | 23.3 | 30.1 | d(Ir)+π(mhtz)+π(npzpy) | |
| 142 | -9.743 | 9.9 | 43.1 | 47.0 | π(mhtz)+π(npzpy) | |
| 4 | 212 | -3.220 | 1.2 | 97.7 | 1.1 | π*(ptaz) |
| 211(L) | -3.309 | 0.9 | 97.0 | 2.1 | π*(ptaz) | |
| 210(H) | -7.379 | 43.7 | 46.7 | 9.6 | d(Ir)+π(ptaz) | |
| 209 | -7.677 | 28.8 | 66.4 | 4.8 | d(Ir)+π(ptaz) | |
| 206 | -8.340 | 21.1 | 53.3 | 25.6 | d(Ir)+π(ptaz)+π(bicb) | |
| 203 | -8.862 | 3.9 | 78.7 | 17.3 | π(ptaz)+π(bicb) | |
Table 5 Molecular orbital compositions in the ground state for complexes 1—4 at DFT/B3LYP level
| Complex | Orbital | Energy/eV | Composition(%) | Main bond type | ||
|---|---|---|---|---|---|---|
| Ir | C^N | A^A | ||||
| 1 | 204 | -3.390 | 1.7 | 93.9 | 4.4 | π*(ptaz) |
| 203 | -3.597 | 1.1 | 2.8 | 96.1 | π*(pzpy) | |
| 202(L) | -4.080 | 3.9 | 2.5 | 93.6 | π*(pzpy) | |
| 201(H) | -7.516 | 42.0 | 54.3 | 3.6 | d(Ir)+π(ptaz) | |
| 200 | -7.777 | 28.6 | 67.3 | 4.2 | d(Ir)+π(ptaz) | |
| 196 | -8.619 | 4.1 | 51.5 | 44.4 | π(ptaz)+π(pzpy) | |
| 2 | 216 | -3.180 | 1.6 | 97.4 | 0.9 | π*(ptaz) |
| 215 | -3.288 | 1.4 | 95.4 | 3.2 | π*(ptaz) | |
| 214(L) | -3.625 | 2.2 | 2.6 | 95.1 | π*(npzpy) | |
| 213(H) | -7.279 | 44.1 | 52.2 | 3.7 | d(Ir)+π(ptaz) | |
| 210 | -8.083 | 19.4 | 53.0 | 27.6 | d(Ir)+π(ptaz)+π(npzpy) | |
| 206 | -8.754 | 0.8 | 98.4 | 0.8 | π(ptaz) | |
| 3 | 153 | -3.339 | 3.2 | 88.5 | 8.3 | π*(mhtz) |
| 150(L) | -4.296 | 3.0 | 2.2 | 94.9 | π*(npzpy) | |
| 149(H) | -7.762 | 43.4 | 48.9 | 7.7 | d(Ir)+π(mhtz) | |
| 148 | -8.149 | 46.7 | 23.3 | 30.1 | d(Ir)+π(mhtz)+π(npzpy) | |
| 142 | -9.743 | 9.9 | 43.1 | 47.0 | π(mhtz)+π(npzpy) | |
| 4 | 212 | -3.220 | 1.2 | 97.7 | 1.1 | π*(ptaz) |
| 211(L) | -3.309 | 0.9 | 97.0 | 2.1 | π*(ptaz) | |
| 210(H) | -7.379 | 43.7 | 46.7 | 9.6 | d(Ir)+π(ptaz) | |
| 209 | -7.677 | 28.8 | 66.4 | 4.8 | d(Ir)+π(ptaz) | |
| 206 | -8.340 | 21.1 | 53.3 | 25.6 | d(Ir)+π(ptaz)+π(bicb) | |
| 203 | -8.862 | 3.9 | 78.7 | 17.3 | π(ptaz)+π(bicb) | |
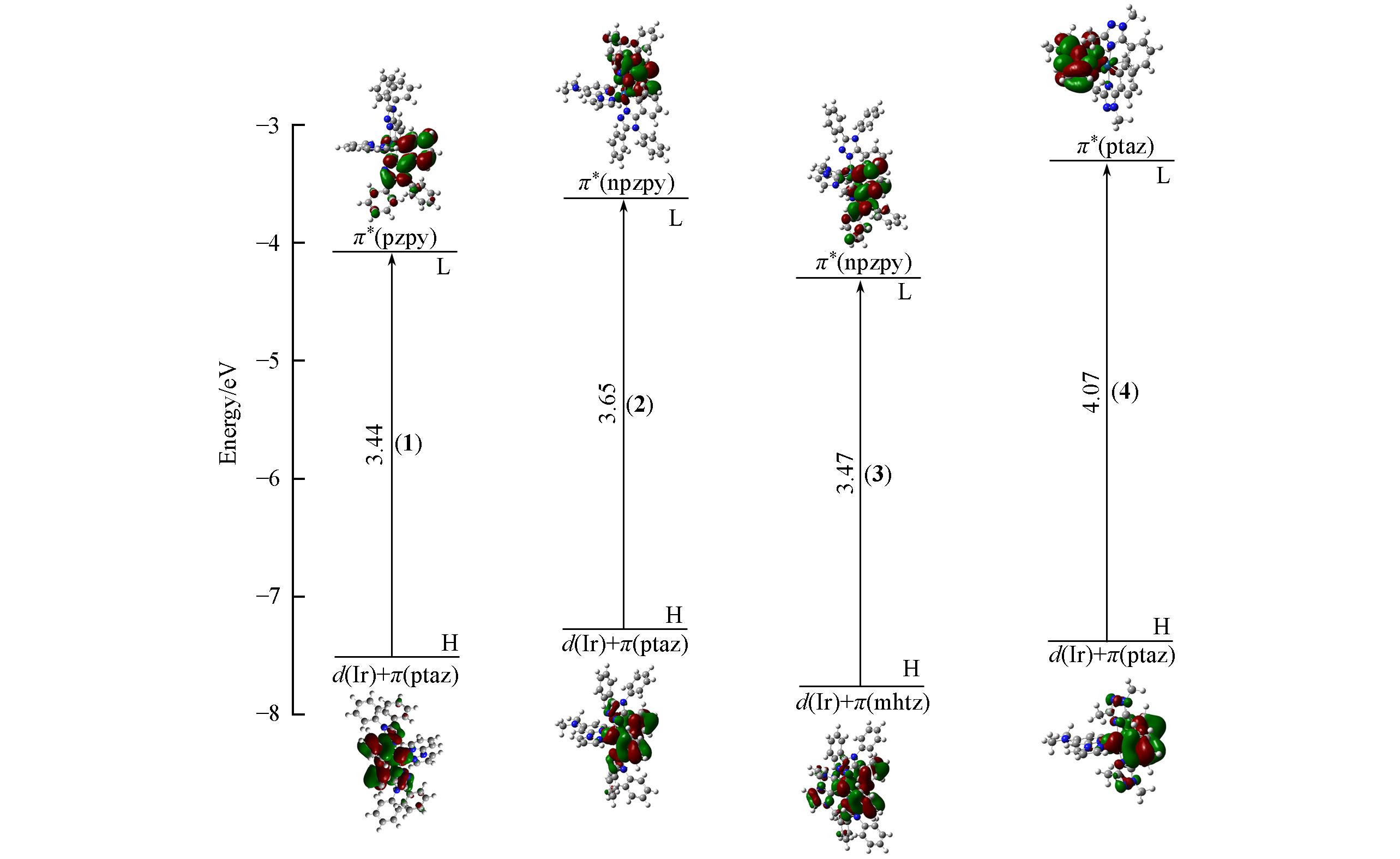
Fig.3 Single electron transition with the lowest energy absorption of complexes 1—4 in CH3CN
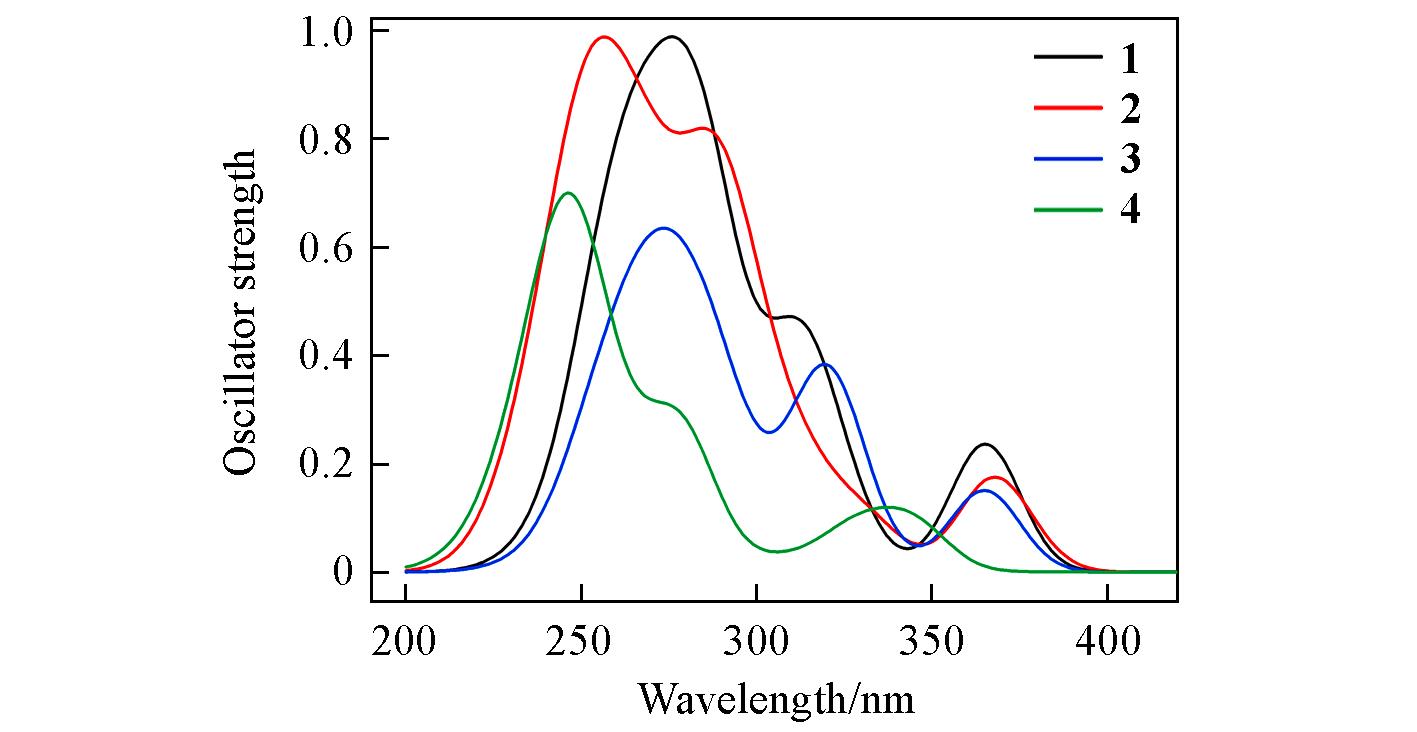
Fig.4 Simulated absorption spectra of complexes 1—4 in CH3CN
| Complex | λmax/nm(Emax/eV) | Configuration | Assignments | Expt. |
|---|---|---|---|---|
| 1 | 503(2.46) | 202→201(0.45) | 3MLCT/3LLCT | 486[ |
| 2 | 506(2.45) | 214→213(0.43) | 3MLCT/3LLCT | 489[ |
| 3 | 468(2.65) | 150→149(0.49) | 3MLCT/3LLCT | 488[ |
| 4 | 511(2.43) | 211→210(0.45) | 3MLCT/3ILCT | 487[ |
Table 6 Phosphorescence spectra of complexes 1—4 in CH3CN at CIS level and experimental values
| Complex | λmax/nm(Emax/eV) | Configuration | Assignments | Expt. |
|---|---|---|---|---|
| 1 | 503(2.46) | 202→201(0.45) | 3MLCT/3LLCT | 486[ |
| 2 | 506(2.45) | 214→213(0.43) | 3MLCT/3LLCT | 489[ |
| 3 | 468(2.65) | 150→149(0.49) | 3MLCT/3LLCT | 488[ |
| 4 | 511(2.43) | 211→210(0.45) | 3MLCT/3ILCT | 487[ |
| Complex | 10-5kr/s-1 | 10-5knr/s-1 | 〈T1|HSOC|S0〉/cm‒1 | Ereor./cm‒1 | Φ | Φp,theory | ||
|---|---|---|---|---|---|---|---|---|
| Exp.[ | Theory | Exp.[ | Theory | |||||
| 1 | 1.0 | 1.15 | 49 | 31.6 | 325 | 2583 | 0.02 | 0.035 |
| 2 | 1.1 | 1.92 | 7.3 | 7.17 | 299 | 1232 | 0.13 | 0.211 |
| 3 | — | 0.47 | — | 2310 | 276 | 4569 | — | 0.000203 |
| 4 | 0.7 | 0.60 | 1.1 | 0.81 | 263 | 975 | 0.37 | 0.426 |
Table 7 Experimental and calculated radiative decay constants kr, nonradiative decay constants knr, 〈T1|Hsoc|S0〉 coupling, the reorganization energy, and the internal quantum efficiency values of complexes 1—4
| Complex | 10-5kr/s-1 | 10-5knr/s-1 | 〈T1|HSOC|S0〉/cm‒1 | Ereor./cm‒1 | Φ | Φp,theory | ||
|---|---|---|---|---|---|---|---|---|
| Exp.[ | Theory | Exp.[ | Theory | |||||
| 1 | 1.0 | 1.15 | 49 | 31.6 | 325 | 2583 | 0.02 | 0.035 |
| 2 | 1.1 | 1.92 | 7.3 | 7.17 | 299 | 1232 | 0.13 | 0.211 |
| 3 | — | 0.47 | — | 2310 | 276 | 4569 | — | 0.000203 |
| 4 | 0.7 | 0.60 | 1.1 | 0.81 | 263 | 975 | 0.37 | 0.426 |
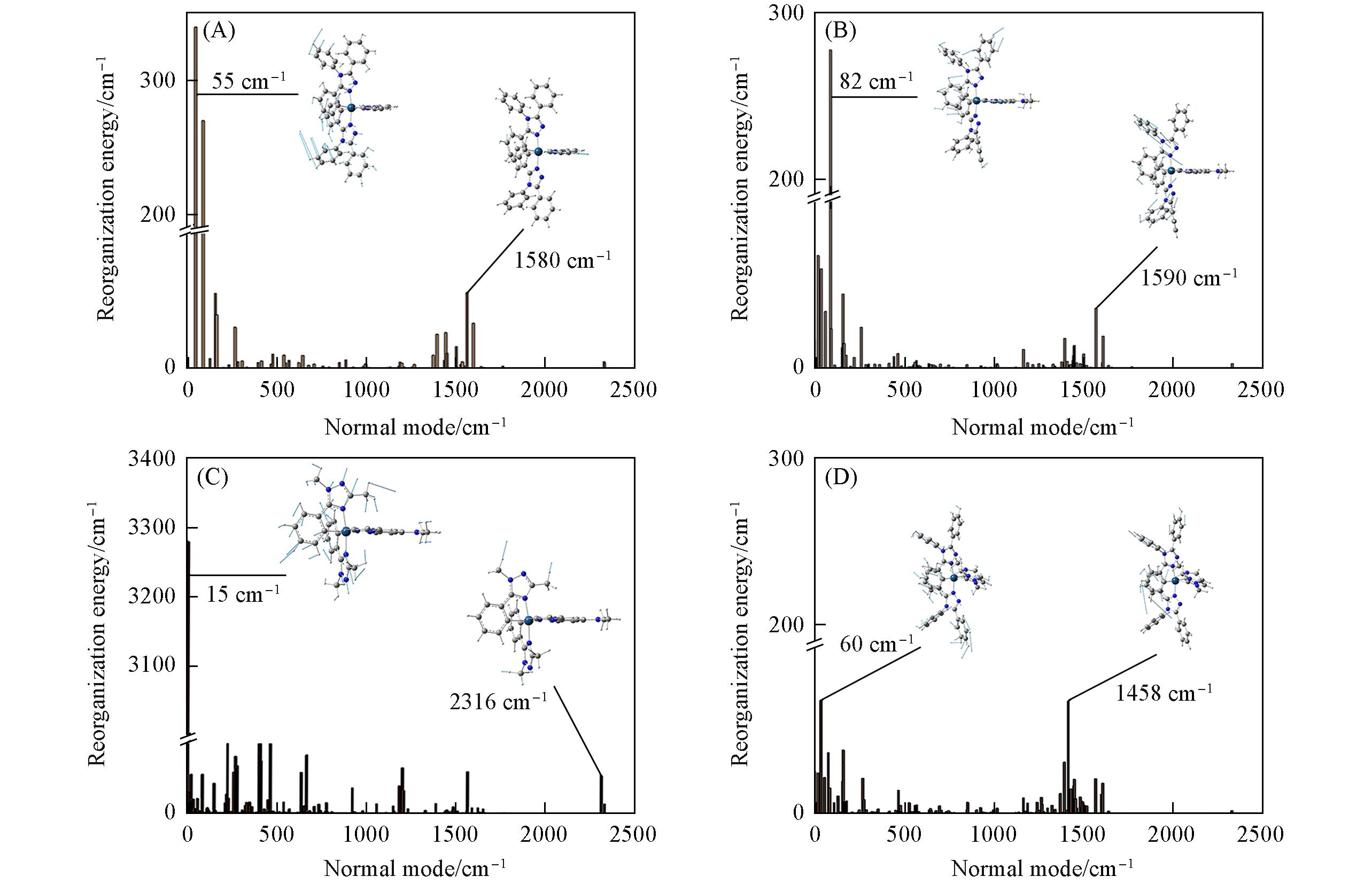
Fig.5 Calculated reorganization energies versus the normal mode frequencies for complexes 1—4Insets are normal-mode displacement vectors for the largest reorganization energy in the low-frequency and high-frequency region.
| 1 | Du H., Akakuru O. U., Yao C., Fang Y., Wu A., Transl. Oncol., 2021, 15(1), 101264 |
| 2 | Somayeh T., Zahra D., Fariba G. N., Hadi B., Peyman M. J., Antonio D. B., Biosens. Bioelectron., 2022, 216, 114674 |
| 3 | Li Y. Y., Gan P. F., Jiang R. H., Zhao Z. W., Ye J. Y., Liu W., Tong M. P., Liang J. L., Sci. Total Environ., 2021, 787, 147626 |
| 4 | Costa R. D., Orti E., Bolink H. J., Monti F., Accorsi G., Armaroli N., Angew. Chem. Int. Ed., 2012, 51(33), 8178—8211 |
| 5 | Bai R. B., Meng X. W., Wang X. X., He L., Adv. Funct. Mater., 2020, 30(33), 1907169 |
| 6 | Ma D., Tsuboi T., Qiu Y., Duan L., Adv. Mater., 2017, 29(3), 1603253 |
| 7 | Fernandez⁃Hernandez J. M., Yang C. H., Beltran J. I., Lemaur V., Polo F., Frohlich R., Cornil J., Cola L. D., J. Am. Chem. Soc., 2011, 133(27), 10543—10558 |
| 8 | Kim K. H., Moon C. K., Lee J. H., Kim S. Y., Kim J. J., Adv. Mater., 2014, 26(23), 3844—3847 |
| 9 | Yu T., Bao Y., Zhao Y., Zhang H., Xu Z., Wu X., J. Organomet. Chem., 2016, 825/826, 33—40 |
| 10 | Lamansky S., Djurovich P., Murphy D., Abdel⁃Razzap F., Thompson M. E., J. Am. Chem. Soc., 2001, 123(18), 4304—4312 |
| 11 | Cho W., Sarada G., Park J. S., Gal Y. S., Lee J. H., Jin S. H., Org. Electron., 2014, 15(10), 2328—2336 |
| 12 | Lam W. H., Lam E. S. H., Yam V. W. W., J. Am. Chem. Soc., 2013, 135(40), 15135—15143 |
| 13 | Peng Q., Shi Q. H., Niu Y. L., Yi Y. P., Sun S. R., Li W. Q., Shuai Z. G., J. Mater. Chem. C, 2016, 4(28), 6829—6838 |
| 14 | Fan J. Z., Zhang Y. C., Zhang K., Liu J., Jiang G. Y., Lin L. L., Wang C. K., Org. Electron., 2019, 71(1), 113—122 |
| 15 | Zhang K., Cai L., Fan J. Z., Zhang Y. C., Lin L. L., Wang C. K., Spectrochim. Acta A, 2019, 209, 248—255 |
| 16 | Wang X. L., Chen C., Li Y. Y., Ning P., Wu W. P., Wang L., Org. Electron., 2017, 49, 360—367 |
| 17 | Shi Q. H., Peng Q., Sun S. R., Shuai Z. G., Acta Chim. Sinica, 2013, 71, 884—891 |
| 18 | Tian M., Yu R., Chen M., He L., Dyes Pigments, 2021, 193(1), 109477 |
| 19 | Wang X., Wang S., Pan F., He L., Duan L., Inorg. Chem., 2019, 58(18), 12132—12145 |
| 20 | Hay P. J., Wadt W. R., J. Chem. Phys., 1985, 82(1), 299—310 |
| 21 | Petersson G. A., Bennett A., Tensfeldt T. G., Al⁃Laham M. A., Shirley W. A., Mantzaris J., J. Chem. Phys., 1988, 89, 2193—2218 |
| 22 | Barone V., Cossi V., Tomasi J., J. Chem. Phys., 1997, 107, 3210—3221 |
| 23 | Lee C., Yang W., Parr R. G., Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785—789 |
| 24 | Zhao Y., Truhlar D. G., J. Phys. Chem. A, 2006, 110, 5121—5129 |
| 25 | Zhao Y., Truhlar D. G., Theor. Chem. Acc., 2008, 120, 215—241 |
| 26 | Yanai T., Tew D. P., Handy N. C., Chem. Phys. Lett., 2004, 393, 51—57 |
| 27 | Bak K. L., Jorgensen P., Helgaker T., Ruud K., Jensen H. J. R. A., J. Chem. Phys., 1993, 98, 8873—8887 |
| 28 | Foresman J. B., Head⁃Gordon M., Pople J. A., J. Phys. Chem., 1992, 96, 135—149 |
| 29 | Liu T., Zhang H. X., Shu X., Xia B. H., Dalton Trans., 2007, 19, 1922—1928 |
| 30 | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Zakrzewsk V. G., Montgomery J. A. Jr., Stratmann R. E., Burant J. C., Dapprich S., Millam J. M., Daniels A. D., Kudin K. N., Strain M. C., Farkas O., Tomasi J., Barone V., Cossi M., Cammi R., Mennucci B., Pomelli C., Adamo C., Clifford S., Ochterski J., Petersson G. A., Ayala P. Y., Cui Q., Morokuma K., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Cioslowski J., Ortiz J. V., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Gomperts R., Martin R. L., Fox D. J., Keith T., Al⁃Laham M. A., Peng C. Y., Nanayakkara A., Gonzalez C., Challacombe M., Gill P. M. W., Johnson B. G., Chen W., Wong M. W., Andres J. L., Head⁃Gordon M., Replogle E. S., Pople J. A., Gaussian 09, Revision D. 02, Gaussian Inc., Wallingford CT, 2009 |
| 31 | Tevelde G. T., Bickelhaupt F. M., Baerends J., Guerra C. F., Ziegler T. J., J. Comput. Chem., 2001, 22(9), 931—967 |
| 32 | Chong D. P., Mol. Phys., 2005, 103(6—8), 749—761 |
| 33 | Smith A. R. G., Burn P. L., Powell B. J., Chem. Phys. Chem., 2011, 12(13), 2428—2437 |
| 34 | Zhang Y., Zhang Z. X., Wang Y., Li H., Bai F. Q., Zhang H. X., Inorg. Chem. Front., 2018, 5, 1016—1025 |
| 35 | Wang Y., Bao P., Wang J., Jia R., Bai F. Q., Zhang H. X., Inorg. Chem., 2018, 57(11), 6561—6570 |
| 36 | Reimers J. R., J. Chem. Phys., 2001, 115(20), 9103—9109 |
| 37 | Wang S. P., Li Y., Zhang Z. X., Zhang Y., Wang Y., Kong S. M., Li H. C., Jian W., Bai F. Q., Zhang H. X., Inorg. Chem., 2021, 60, 1480—1490 |
| [1] | 王霄靖, 刘艺霞, 李阳, 杨陈宗, 冯敏强, 樊健. 基于四氰基受体单元的高效近红外热激活延迟荧光[J]. 高等学校化学学报, 2023, 44(12): 20230274. |
| [2] | 夏天, 万家炜, 于然波. 异原子配位结构碳基单原子电催化剂结构与性能相关性的研究进展[J]. 高等学校化学学报, 2022, 43(5): 20220162. |
| [3] | 王祖民, 孟程, 于然波. 过渡金属磷化物析氢催化剂的掺杂调控[J]. 高等学校化学学报, 2022, 43(11): 20220544. |
| [4] | 卓增庆, 潘锋. 基于软X射线光谱的锂电池材料的电子结构与演变的研究进展[J]. 高等学校化学学报, 2021, 42(8): 2332. |
| [5] | 王坤华, 姚纪松, 杨俊楠, 宋永慧, 刘雨莹, 姚宏斌. 金属卤化物钙钛矿纳米晶高效发光二极管的制备与器件性能优化[J]. 高等学校化学学报, 2021, 42(5): 1464. |
| [6] | 史海涵,吴香萍,彭辛哲,余国静,董朝阳,纪瑶瑶,杨思文,陈俊林,王锦,冉雪芹,杨磊,解令海,黄维. 一种基于风车格结构的有效降低内重组能的咔唑类格子化分子[J]. 高等学校化学学报, 2020, 41(7): 1670. |
| [7] | 孙国栋, 王雪, 江国亮, 徐之勇, 刘洪梅. 二维金属-六亚氨基苯框架材料的气体吸附效应[J]. 高等学校化学学报, 2019, 40(5): 995. |
| [8] | 周和根, 金华, 郭辉瑞, 林晶, 章永凡. 黄铜矿型铜基硫属半导体材料的电子结构和光学性质[J]. 高等学校化学学报, 2019, 40(3): 518. |
| [9] | 张兆燕,陈宏善. Al6ONa2组装Zintl相晶体的理论研究[J]. 高等学校化学学报, 2019, 40(11): 2354. |
| [10] | 田琳飞, 张春华, 曲宁, 毕艳婷, 张红星, 潘清江. 双层三明治四聚吡咯铀配合物的结构设计和稳定性理论计算[J]. 高等学校化学学报, 2018, 39(4): 749. |
| [11] | 刘豫龙, 路芳, 路萍. 基于芘并咪唑的电致发光材料的合成与表征[J]. 高等学校化学学报, 2017, 38(4): 583. |
| [12] | 李坦, 张小超, 王凯, 李瑞, 樊彩梅. α,β,γ,δ,ε,η-Bi2O3电子结构和光学性质的第一性原理研究[J]. 高等学校化学学报, 2016, 37(5): 920. |
| [13] | 仓玉萍, 陈东, 杨帆, 杨慧明. 氮化锗多形体的四方、 单斜和正交畸变的理论研究[J]. 高等学校化学学报, 2016, 37(4): 674. |
| [14] | 欧阳巧凤, 郑兰兰, 曹红, 张馨, 李春, 沈姣, 陈礼婷, 刘进. 离子液体[MBPy]Tf2N的光谱特征及荧光法检测[J]. 高等学校化学学报, 2015, 36(10): 1906. |
| [15] | 李维军, 高曌, 王志明, 杨兵, 路萍, 马於光. 含菲并咪唑基团的蓝色电致发光材料[J]. 高等学校化学学报, 2014, 35(9): 1849. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||