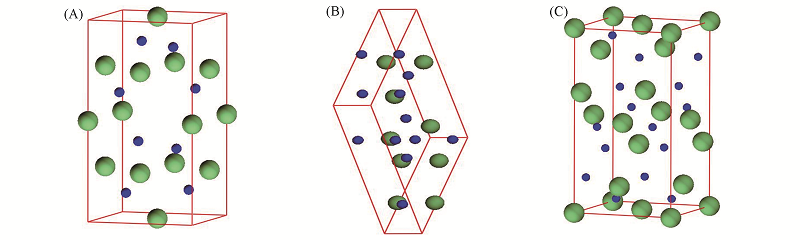
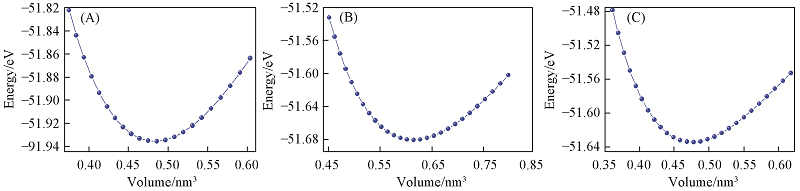
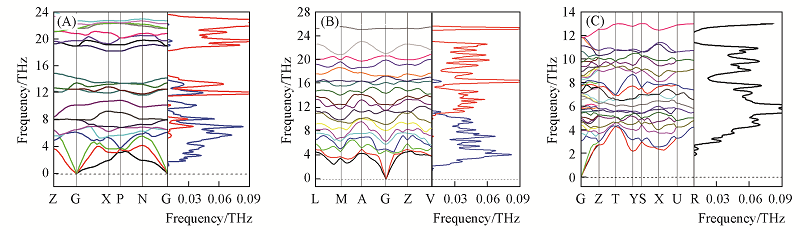
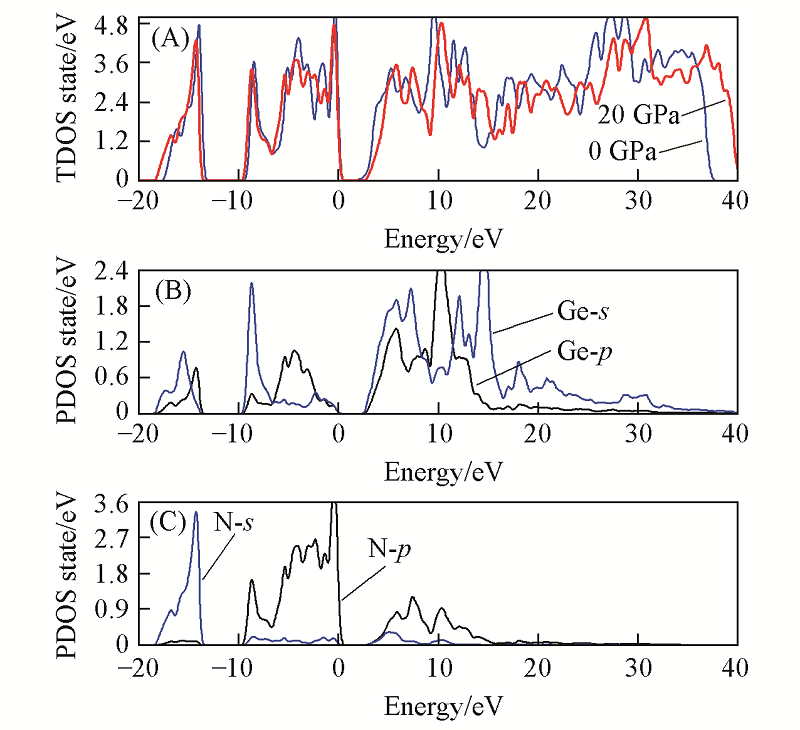
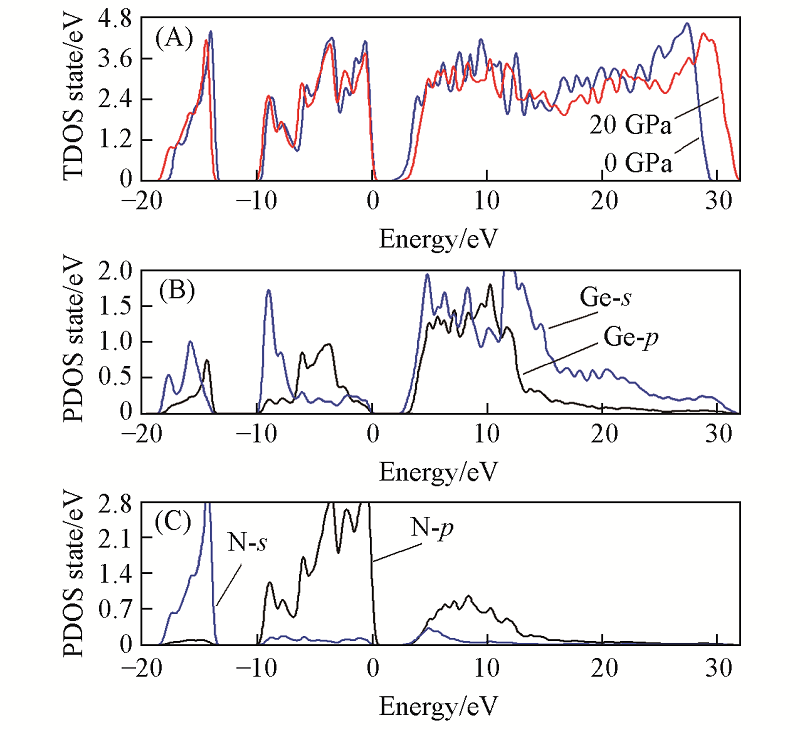
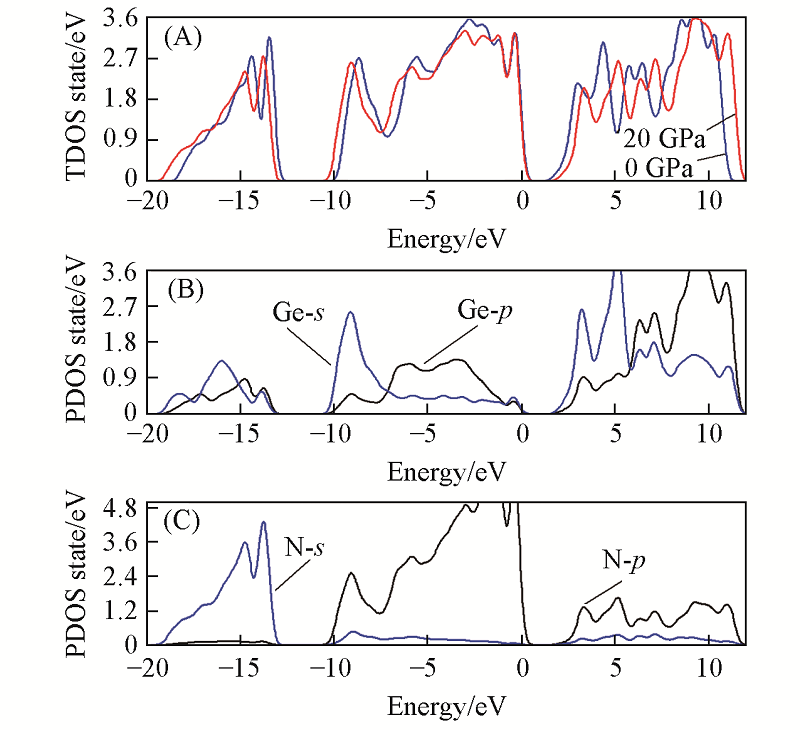
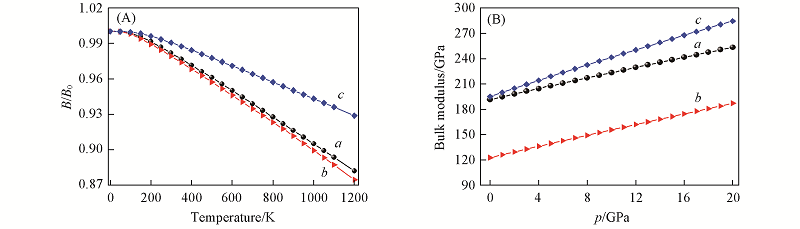
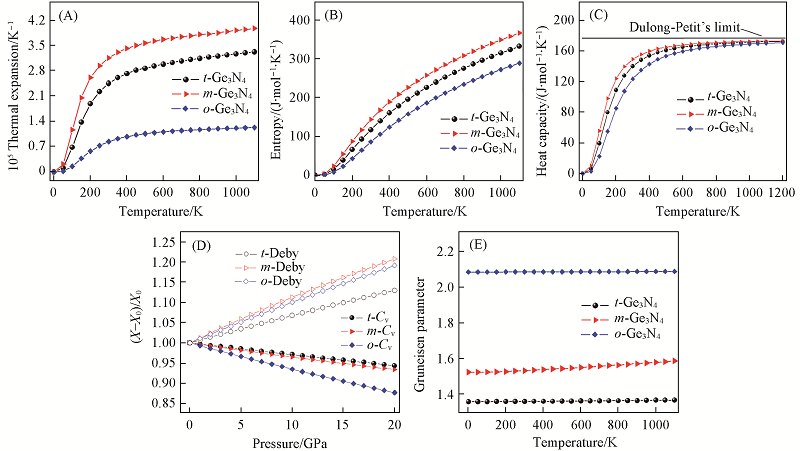
| [1] |
Lu X., La P., Guo X., Wei Y., Nan X., He L., J. Phys. Chem. Lett., 2013, 4(11), 1878—1881
|
| [2] |
Fu R. B., Yang L. Q., Feng L. Y., Guo W., Chem. J. Chinese Universities, 2014, 35(4), 825—830
|
|
(付融冰, 杨兰琴, 冯雷雨, 郭伟. 高等学校化学学报, 2014, 35(4), 825—830)
|
| [3] |
Hart J. N., Allan N. L., Claeyssens F., Phys. Rev. B, 2011, 84(24), 245209
|
| [4] |
Yang H. M., Xu B., Guo X. F., Wen Z. X., Fan X. H., Tian B., Chem. J. Chinese Universities, 2013, 34(4), 970—974
|
|
(杨红梅, 许斌, 郭晓斐, 温振兴, 范小红, 田彬. 高等学校化学学报, 2013, 34(4), 970—974)
|
| [5] |
Salamat A., Hector A. L., Kroll P., McMillan P. F., Coordin. Chem. Rev., 2013, 257(13/14), 2063—2072
|
| [6] |
Ding Y. C., Chen M., Wu W. J., J. Theor. Comput. Chem., 2015, 14(4), 1550024
|
| [7] |
Boyko T. D., Hunt A., Zerr A., Moewes A., Phys. Rev. Lett., 2013, 111(9), 097402
|
| [8] |
Deb S. K., Dong J., Hubert H., McMillan P. F., Sankey O. F., Solid State Commun., 2000, 114(3), 137—142
|
| [9] |
Duan Y. H., Zhang K. M., Xie X. D., Acta Phys. Sinica, 1996, 45(3), 512—517
|
|
(段玉华, 张开明, 谢希德. 物理学报, 1996, 45(3), 512—517)
|
| [10] |
Serghiou G., Miehe G., Tschauner O., Zerr A., Boehler R., J. Chem. Phys., 1999, 111(10), 4659—4662
|
| [11] |
Leinenweber K., O’Keeffe M., Somayazulu M. S., Hubert H., McMillan P. F., Wolf G. H., Chem. Eur. J., 1999, 5(10), 3076—3078
|
| [12] |
Yang M., Wang S. J., Feng Y. P., Peng G. W., Sun Y. Y., J. Appl. Phys., 2007, 102(1), 013507
|
| [13] |
Chu L. H., Kozhevnikov A., Schulthess T. C., Cheng H. P., J. Chem. Phys., 2014, 141(4), 044709
|
| [14] |
He H. L., Sekine T., Kobayashi T., Kimoto K., J. Appl. Phys., 2001, 90(9), 4403—4406
|
| [15] |
Wang Z., Zhao Y., Schiferl D., Qian J., Downs R. T., Mao H. K., Sekine T., J. Phys. Chem. B, 2013, 107(51), 14151—14153
|
| [16] |
Dong J. J., Sankey O. F., Deb S. K., Wolf G., McMillan P. F., Phys. Rev. B, 2000, 61(18), 11979—11992
|
| [17] |
Gao S. P., Cai G. H., Xu Y., Comput. Mater. Sci., 2013, 67, 292—295
|
| [18] |
Lü T. Y., Zheng J. C., Chem. Phys. Lett., 2010, 501(1—3), 47—53
|
| [19] |
Wang H., Chen Y., Kaneta Y., Iwata S., J. Phys.: Condens. Matter., 2006, 18(47), 10663—10676
|
| [20] |
Ching W. Y., Mo S. D., Ouyang L. Z., Phys. Rev. B, 2001, 63(24), 245110
|
| [21] |
Horvath-Bordon E., Riedel R., Zerr A., McMillan P. F., Auffermann G., Prots Y., Bronger W., Kniep R., Kroll P., Chem. Soc. Rev., 2006, 35(10), 987—1014
|
| [22] |
Soignard E., McMillan P. F., Hejny C., Leinenweber K., J. Solid State Chem., 2004, 177(1), 299—311
|
| [23] |
McMillan P. F., Deb S. K., Dong J. J., J. Raman Spectrosc., 2003, 34(7/8), 567—577
|
| [24] |
Luo Y., Cang Y., Chen D., Comput. Condens. Matter., 2014, 1, 1—7
|
| [25] |
Lowther J. E., Phys. Rev. B, 2000, 62(1), 5—8
|
| [26] |
Cui L., Hu M., Wang Q., Xu B., Yu D., Liu Z., He J., J. Solid State Chem., 2015, 228, 20—26
|
| [27] |
Kohn W., Sham L. J., Phys. Rev., 1965, 140(4A), A1133—A1138
|
| [28] |
Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett., 1996, 77(18), 3865—3868
|
| [29] |
Monkhorst H. J., Pack J. D., Phys. Rev. B, 1976, 13(12), 5188—5192
|
| [30] |
Ackland G. J., Warren M. C., Clark S. J., J. Phys.: Condens. Matter., 1997, 9(37), 7861—7872
|
| [31] |
Shanno D. F., Kettler P. C., Math. Comput., 1970, 24(1—3), 657—664
|
| [32] |
Otero-de-la-Roza A., Luańa V., Comput. Phys. Commun., 2011, 182(8), 1708—1720
|
| [33] |
Otero-de-la-Roza A., Abbasi-Pérez D., Luańa V., Comput. Phys. Commun., 2011, 182(10), 2232—2248
|
| [34] |
Dong J. J., Deslippe J., Sankey O. F., Soignard E., McMillan P. F., Phys. Rev. B, 2003, 67(9), 094104
|
| [35] |
Liu Q. J., Ran Z., Liu F. S., Liu Z. T., J. Alloys Compd., 2015, 631, 192—201
|
| [36] |
Soignard E., Somayazulu M., Dong J. J., Sankey O. F., McMillan P. F., J. Phys.: Condens. Matter., 2001, 13(4), 557—563
|
| [37] |
Yu B. H., Chen D., Chin. Phys. B, 2012, 61(19), 197102
|
| [38] |
Zhao Z. F., Li Y. F., Yao N., Chem. J. Chinese Universities, 2015, 36(8), 1648—1654
|
|
(赵昨非, 李元丰, 姚宁. 高等学校化学学报, 2015, 36(8), 1648—1654)
|
| [39] |
Petit A. T., Dulong P. L., Ann. Chim. Phys., 1819, 10, 395—413
|
 ), 杨帆, 杨慧明
), 杨帆, 杨慧明