

Chem. J. Chinese Universities ›› 2015, Vol. 36 ›› Issue (11): 2171.doi: 10.7503/cjcu20150546
• Physical Chemistry • Previous Articles Next Articles
GUO Kai, ZHANG Heng, SUN Jichao, YUAN Shiling*( ), LIU Chengbu
), LIU Chengbu
Received:2015-07-14
Online:2015-11-10
Published:2015-10-12
Contact:
YUAN Shiling
E-mail:shilingyuan@sdu.edu.cn
Supported by:CLC Number:
TrendMD:
GUO Kai, ZHANG Heng, SUN Jichao, YUAN Shiling, LIU Chengbu. Molecular Dynamics Simulation on Self-Assembly of Fmoc-FF Dipeptide†[J]. Chem. J. Chinese Universities, 2015, 36(11): 2171.
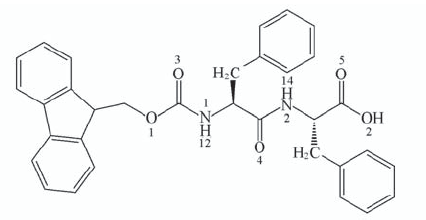
Fig.1 Molecular structure of Fmoc-FF
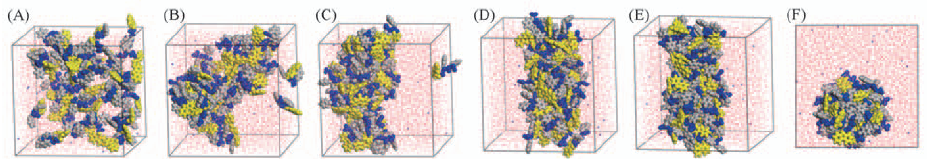
Fig.2 Views of the configurations at different time(A—E) and top view of configuration at 50 ns(F) Time/ns: (A) 0; (B) 1; (C) 5; (D) 30; (E) 50. Orange represents the fluorenyl group of Fmoc, blue is the peptide chain, and gray is the benzene ring of peptide chain. Fmoc-FF molecules are represented by CPK pattern. Water molecules are represented by red and white lines.
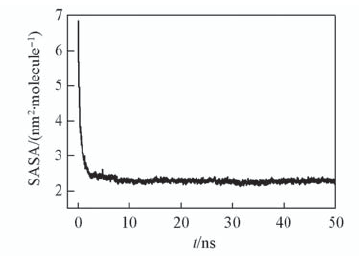
Fig.3 Solvent accessible surface area(SASA) of Fmoc-FF molecule
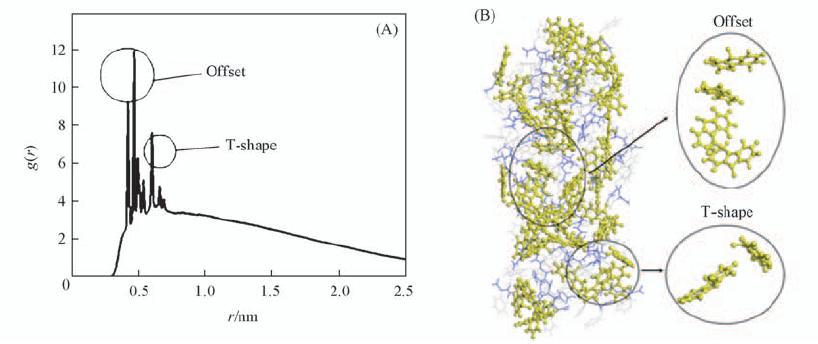
Fig.4 Non-normalized RDF plot of distance between fluorenyl rings(A) and MD simulation of π-π stacking of the aggregate after 50 ns(B) Water molecules are omitted to show the structure clearly.
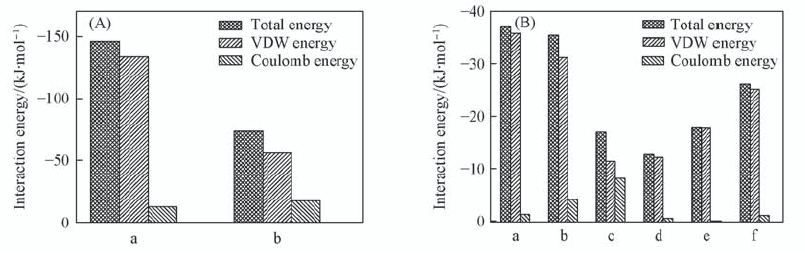
Fig.5 Overall interaction energy and the decomposed VDW, Coulomb and hydrogen bond terms (A) The whole interaction energy of dipeptide and water molecules. a. Peptide-peptide; b. peptide-H2O. (B) The individual interaction energy between different parts of Fmoc-FF dipeptide. a. Fmoc-Fmoc; b. Fmoc-Chain; c. Chain-Chain; d. Ben-Ben; e. Ben-Chain; f. Ben-Fmoc. All data are averaged using the last 20 ns MD simulations.
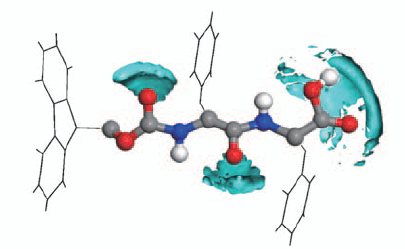
Fig.6 Spatial distribution function between Fmoc-FF molecule and water molecules
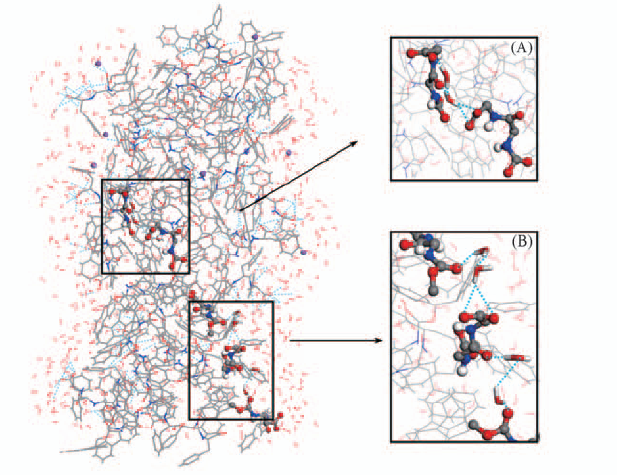
Fig.7 Views of water bridge structure (A) Dipeptides formed water bridge through one water molecule; (B) dipeptides formed water bridge by two water molecules.
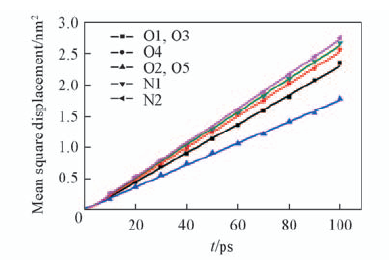
Fig.8 Mean square displacement-time curves of water around certain atoms of Fmoc-FF dipeptide
| Certain atoms of Fmoc-FF dipeptide | 105Diffusion coefficient(cm2·s-1) | Residence time, τr/ps |
|---|---|---|
| O1, O3 | 3.87 | 7.09 |
| O4 | 4.27 | 9.52 |
| O2, O5 | 2.91 | 10.65 |
| N1 | 4.42 | 1.71 |
| N2 | 4.58 | 1.43 |
Table 1 Dynamic properties of water around certain atoms of Fmoc-FF dipeptide
| Certain atoms of Fmoc-FF dipeptide | 105Diffusion coefficient(cm2·s-1) | Residence time, τr/ps |
|---|---|---|
| O1, O3 | 3.87 | 7.09 |
| O4 | 4.27 | 9.52 |
| O2, O5 | 2.91 | 10.65 |
| N1 | 4.42 | 1.71 |
| N2 | 4.58 | 1.43 |
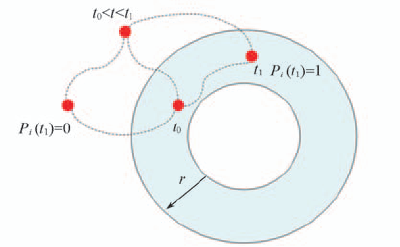
Fig.9 Illustration of definition of Pi Pi(t1)=1 when water molecule in the first shell at t0 and t1.The location of water molecule during t0—t1 does not count.
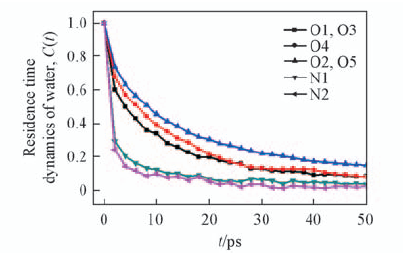
Fig.10 Time correlation functions of water around certain atoms of Fmoc-FF dipeptide
| Atom type | Number of hydrogen bonds | Residence time/ps |
|---|---|---|
| O1, O3 | 12.68 | 16.53 |
| N1H12 | 0.92 | 2.23 |
| O4 | 22.77 | 12.56 |
| N2H14 | 1.61 | 1.01 |
| O2, O5 | 105.82 | 9.54 |
Table 2 Averaged hydrogen bond number and residence time between certain atoms of Fmoc-FF dipeptide and water molecules
| Atom type | Number of hydrogen bonds | Residence time/ps |
|---|---|---|
| O1, O3 | 12.68 | 16.53 |
| N1H12 | 0.92 | 2.23 |
| O4 | 22.77 | 12.56 |
| N2H14 | 1.61 | 1.01 |
| O2, O5 | 105.82 | 9.54 |
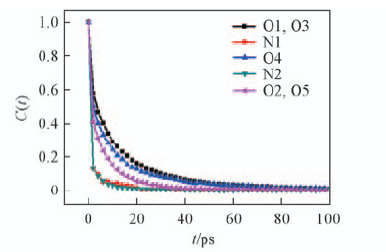
Fig.11 Time correlation function[C(t)] for the hydrogen bonds formed between certain atoms of Fmoc-FF dipeptide and water molecules
| [1] | Peppas N. A., Bures P., Leobandung W., Ichikawa H., Eur. J. Pharm. Biopharm., 2000, 50(1), 27—46 |
| [2] | Peppas N. A., Hilt J. Z., Khademhosseini A., Langer R., Adv. Mater., 2006, 18(11), 1345—1360 |
| [3] | Slaughter B. V., Khurshid S. S., Fisher O. Z., Khademhosseini A., Peppas N. A., Adv. Mater., 2009, 21(32/33), 3307—3329 |
| [4] | Hoffman A. S., Adv. Drug Deliv. Rev., 2002, 54(1), 3—12 |
| [5] | Drury J. L., Mooney D. J., Biomaterials, 2003, 24(24), 4337—4351 |
| [6] | Schneider J. P., Pochan D. J., Ozbas B., Rajagopal K., Pakstis L., Kretsinger J., J. Am. Chem. Soc., 2002, 124(50), 15030—15037 |
| [7] | Reches M., Gazit E., Israel J. Chem., 2005, 45(3), 363—371 |
| [8] | Guo C., Luo Y., Zhou R., Wei G., ACS Nano, 2012, 6(5), 3907—3918 |
| [9] | Jiang S., Cao Z., Adv. Mater., 2010, 22(9), 920—932 |
| [10] | Zhou M., Smith A. M., Das A. K., Hodson N. W., Collins R. F., Ulijn R. V., Gough J. E., Biomaterials, 2009, 30(13), 2523—2530 |
| [11] | Mahler A., Reches M., Rechter M., Cohen S., Gazit E., Adv. Mater., 2006, 18(11), 1365—1370 |
| [12] | Silva G. A., Czeisler C., Niece K. L., Beniash E., Harrington D. A., Kessler J. A., Stupp S. I., Science, 2004, 303(5662), 1352—1355 |
| [13] | Chen L., Morris K., Laybourn A., Elias D., Hicks M. R., Rodger A., Serpell L., Adams D. J., Langmuir, 2010, 26(7), 5232—5242 |
| [14] | Chen L., Revel S., Morris K., Serpell C. L., Adams D. J., Langmuir, 2010, 26(16), 13466—13471 |
| [15] | Huang Y., Qiu Z., Xu Y., Shi J., Lin H., Zhang Y., Org. Biomol. Chem., 2011, 9(7), 2149—2155 |
| [16] | Ma M., Kuang Y., Gao Y., Zhang Y., Gao P., Xu B., J. Am. Chem. Soc., 2010, 132(8), 2719—2728 |
| [17] | Smith A. M., Williams R. J., Tang C., Coppo P., Collins R. F., Turner M. L., Saiani A., Ulijn R. V., Adv. Mater., 2008, 20(1), 37—41 |
| [18] | Orbach R., Adler-Abramovich L., Zigerson S., Mironi-Harpaz I., Seliktar D., Gazit E., Biomacromolecules, 2009, 10(9), 2646—2651 |
| [19] | Orbach R., Mironi-Harpaz I., Adler-Abramovich L., Mossou E., Mitchell E. P., Forsyth V. T., Gazit E., Seliktar D., Langmuir, 2012, 28(4), 2015—2022 |
| [20] | Hirst A. R., Roy S., Arora M., Das A. K., Hodson N., Murray P., Marshall S., Javid N., Sefcik J., Boekhoven J., van Esch J. H., Santabarbara S., Hunt N. T., Ulijn R. V., Nat. Chem., 2010, 2(12), 1089—1094 |
| [21] | Jayawarna V., Richardson S. M., Hirst A. R., Hodson N. W., Saiani A., Gough J. E., Ulijn R. V., Acta Biomater., 2009, 5(3), 934—943 |
| [22] | Debnath S., Shome A., Das D., Das P. K., J. Phys. Chem. B, 2010, 114(13), 4407—4415 |
| [23] | Tang C., Smith A. M., Collins R. F., Ulijn R. V., Saiani A., Langmuir, 2009, 25(16), 9447—9453 |
| [24] | Tamamis P., Adler-Abramovich L., Reches M., Marshall K., Sikorski P., Serpell L., Gazit E., Archontis G., Biophys. J., 2009, 96(12), 5020—5029 |
| [25] | Jeon J., Mills C. E., Shell M. S., J. Phys. Chem. B, 2013, 117(15), 3935—3943 |
| [26] | Mu X., Eckes K. M., Nguyen M. M., Suggs L. J., Ren P., Biomacromolecules, 2012, 13(11), 3562—3571 |
| [27] | Malde A. K., Zuo L., Breeze M., Stroet M., Poger D., Nair P. C., Oostenbrink C., Mark A. E, J. Chem. Theory Comput., 2011, 7(12), 4026—4037 |
| [28] | Hess B., Kutzner C., van Der Spoel D., Lindahl E., J. Chem. Theory Comput., 2008, 4(3), 435—447 |
| [29] | Berendsen H.J. C., Postma J. P. M., van Gunsteren W. F., Hermans J., Intermolecular Forces, Reidel, Dordrecht, 1981 |
| [30] | Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J., J. Chem. Phys., 1984, 81(8), 3684—3690 |
| [31] | Chen Z.L., Xu W. R., Tang L. D., The Theory and Application of Molecular Simulation, Chemical Industry Press, Beijing, 2007, 112—120 |
| (陈正隆, 徐为人, 汤立达. 分子模拟的理论与实践, 北京: 化学工业出版社, 2007, 112—120) | |
| [32] | Shao Q., He Y., White A. D., Jiang S. Y., J. Phys. Chem. B, 2010, 114(49), 16625—16631 |
| [33] | He Y., Hower J., Chen S. F., Bernards M. T., Chang Y., Jiang S. Y., Langmuir, 2008, 24(18), 10358—10364 |
| [34] | Lopez C. F., Nielsen S. O., Klein M. L., Moore P. B., J. Phys. Chem. B, 2004, 108(21), 6603—6610 |
| [1] | WANG Xuebin, XUE Yuan, MAO Hua’nyu, XIANG Yanxin, BAO Chunyan. Preparation of Photo/reduction Dual-responsive Hydrogel Microspheres and Their Application in Three-dimensional Cell Culture [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220116. |
| [2] | GAO Zhiwei, LI Junwei, SHI Sai, FU Qiang, JIA Junru, AN Hailong. Analysis of Gating Characteristics of TRPM8 Channel Based on Molecular Dynamics [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220080. |
| [3] | MIN Jing, WANG Liyan. 1H NMR Study on the Conformation of Aromatic Amides Limited by Three-center Hydrogen Bonds [J]. Chem. J. Chinese Universities, 2022, 43(6): 20220084. |
| [4] | HUANG Yi, LYU Lingling, PAN Xiaopeng, SUN Guangdong, LI Yongqiang, YAO Juming, SHAO Jianzhong. Three-dimensional Printing of Photocrosslinked Self-supporting Silk Fibroin Hydrogels [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210841. |
| [5] | ZHANG Yong, XU Jun, BAO Yu, CUI Shuxun. Quantifying the Degree of Weakening Effect of Nonpolar Organic Solvent on the Strength of Intramolecular Hydrogen Bonding by Single-molecule Force Spectroscopy [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210863. |
| [6] | CUI Shaoli, ZHANG Weijia, SHAO Xueguang, CAI Wensheng. Revealing the Effect of Threonine on the Binding Ability of Antifreeze Proteins with Ice Crystals by Free-energy Calculations [J]. Chem. J. Chinese Universities, 2022, 43(3): 20210838. |
| [7] | HU Bo, ZHU Haochen. Dielectric Constant of Confined Water in a Bilayer Graphene Oxide Nanosystem [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210614. |
| [8] | ZHOU Yonghui, LI Yao, WU Yuxuan, TIAN Jing, XU Longquan, FEI Xu. Synthesis of A Novel Photoluminescence Self-healing Hydrogel [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210606. |
| [9] | ZHANG Mi, TIAN Yafeng, GAO Keli, HOU Hua, WANG Baoshan. Molecular Dynamics Simulation of the Physicochemical Properties of Trifluoromethanesulfonyl Fluoride Dielectrics [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220424. |
| [10] | YAN Shuting, YAO Yuan, TAO Xinfeng, LIN Shaoliang. Synthesis and Properties of Polypeptoid Hydrogels Containing Sulfonium Groups [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220381. |
| [11] | GAO Huiling, CAO Zhenzhen, GU Fang, WANG Haijun. Monte Carlo Simulation on Self-healing Behaviour of Hydrogen-bonded Hydrogel [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220482. |
| [12] | LI Congcong, LIU Minghao, HAN Jiarui, ZHU Jingxuan, HAN Weiwei, LI Wannan. Theoretical Study of the Catalytic Activity of VmoLac Non-specific Substrates Based on Molecular Dynamics Simulations [J]. Chem. J. Chinese Universities, 2021, 42(8): 2518. |
| [13] | CAI Yaqian, ZHANG Jiahuai, LIU Fangzhe, LI Haichao, SHI Jianping, GUAN Shuang. Protein-based Hydrogel Assisted by Hofmeister Effect for Strain Sensor [J]. Chem. J. Chinese Universities, 2021, 42(8): 2609. |
| [14] | LEI Xiaotong, JIN Yiqing, MENG Xuanyu. Prediction of the Binding Site of PIP2 in the TREK-1 Channel Based on Molecular Modeling [J]. Chem. J. Chinese Universities, 2021, 42(8): 2550. |
| [15] | ZENG Yonghui, YAN Tianying. Vibrational Density of States Analysis of Proton Hydration Structure [J]. Chem. J. Chinese Universities, 2021, 42(6): 1855. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||