

高等学校化学学报 ›› 2026, Vol. 47 ›› Issue (6): 20250401.doi: 10.7503/cjcu20250401
钟彬焱1, 冯玄2, 高余福1, 詹森华1, 石彤非1( )
)
收稿日期:2025-12-26
出版日期:2026-06-10
发布日期:2026-04-01
通讯作者:
石彤非
E-mail:tfshi@gdut.edu.cn
基金资助:
ZHONG Binyan1, FENG Xuan2, GAO Yufu1, ZHAN Senhua1, SHI Tongfei1()
Received:2025-12-26
Online:2026-06-10
Published:2026-04-01
Contact:
SHI Tongfei
E-mail:tfshi@gdut.edu.cn
Supported by:摘要:
结合分子对接与分子动力学(MD)模拟方法, 探究了磷酸化三螺旋线性β-(1→3)-葡聚糖(TH-CP)与树突状细胞相关C型凝集素-1(Dectin-1)之间的相互作用机制. 研究结果表明, O6及O4,6位点的磷酸化修饰在Dectin-1表面引入了稳定的潜在结合位点, 使TH-CP能够与Dectin-1形成热力学上有利且结构稳定的复合物. 在潜在位点的3种识别方式中, 呈表位识别模式的复合物表现出显著的结合优势. 这主要源于表位识别构型下, TH-CP能够与潜在位点周围更多的氨基酸残基发生协同作用, 从而获得更强的范德华相互作用和静电相互作用. 此外, 磷酸基团中多个羟基可作为灵活的氢键供体和受体, 构建区别于经典位点的新型氢键网络, 使潜在位点介导的识别能力与未修饰β-(1→3)-葡聚糖在经典结合位点的识别能力相当.
中图分类号:
TrendMD:
钟彬焱, 冯玄, 高余福, 詹森华, 石彤非. 基于分子动力学模拟的潜在识别区域下Dectin-1对磷酸化 β-(1→3)-D-葡聚糖的识别机制. 高等学校化学学报, 2026, 47(6): 20250401.
ZHONG Binyan, FENG Xuan, GAO Yufu, ZHAN Senhua, SHI Tongfei. Recognition Mechanism of Dectin-1 for Phosphorylated β -(1→3)-D-Glucan in Potential Recognition Regions Based on Molecular Dynamics Simulations. Chem. J. Chinese Universities, 2026, 47(6): 20250401.
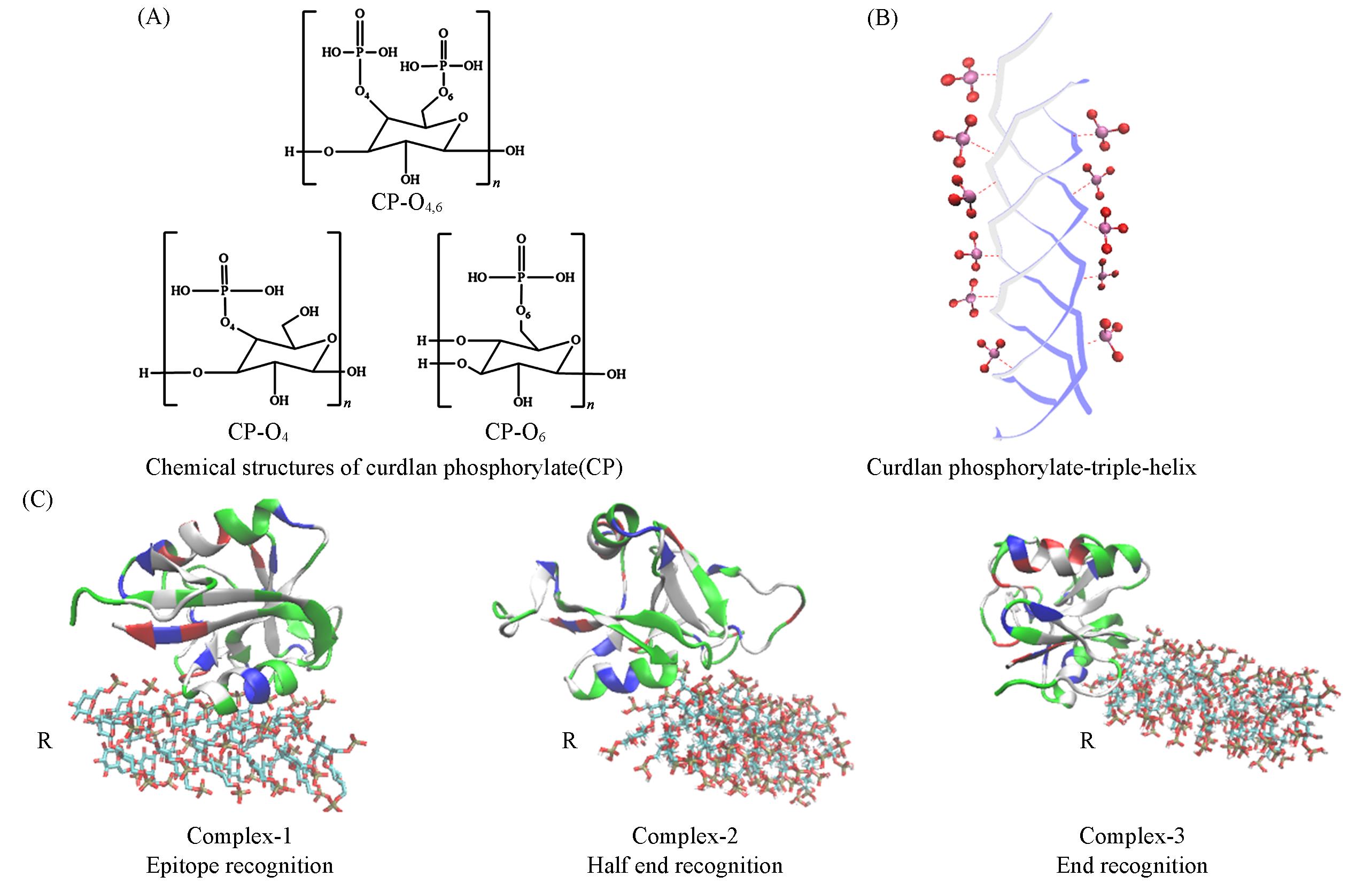
Fig.1 The main discussion focuses on CP and its complex structure(A) Chemical structures showing phosphorylation substitution at the O4, O6 and O4,6 sites; (B) triple helical β-(1→3)-glucan structure with 12 glucose units per chain; oxygen atoms are shown in red and phosphorus atoms in purple; hydrogen atoms are removed for clarity; (C) complexes with three different binding modes, where R denotes the reducing end; molecular models are visualized using the molecular graphics software VMD[42].
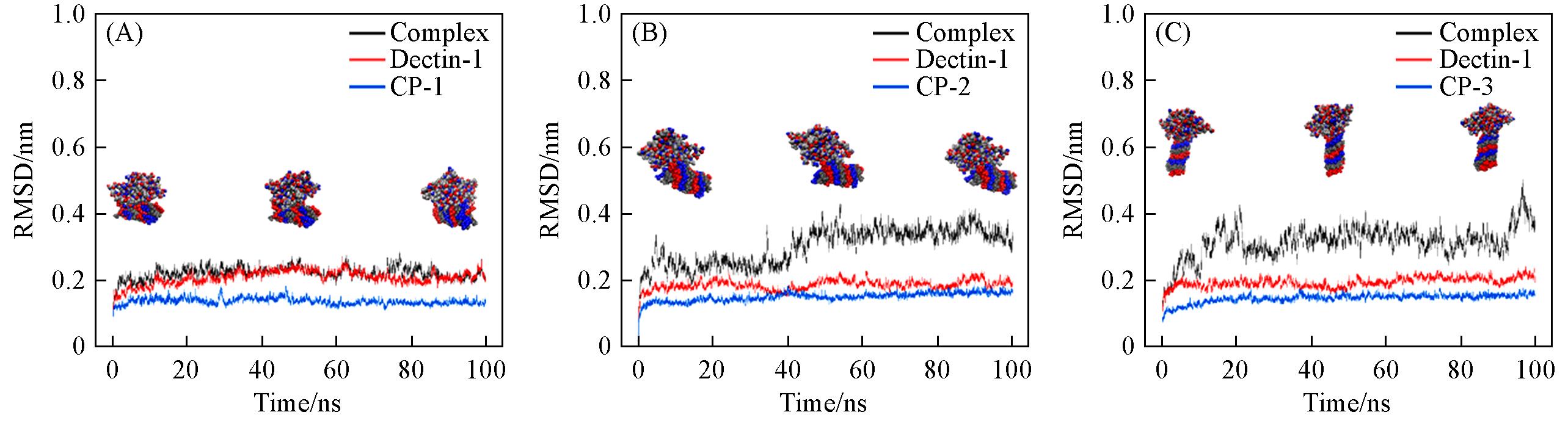
Fig.2 RMSDs of CP⁃1 substituted with O6 and Dectin⁃1 and their corresponding complex(Complex⁃1, A), CP⁃2 substituted with O4,6 and Dectin⁃1 and their corresponding complex(Complex⁃2, B), and CP⁃3 substituted with O4,6 and Dectin⁃1 and their corresponding complex(Complex⁃3, C)The insets show the representative structural snapshots of the complexes at different simulation times.
| Gmx_mmpbsa | ΔEvdW/(kJ·mol-1) | ΔEelec/(kJ·mol-1) | ΔGpolar/(kJ·mol-1) | ΔGnonpolar/(kJ·mol-1) | ΔGbind/(kJ·mol-1) |
|---|---|---|---|---|---|
| TH⁃bglc | -231.681 | -278.322 | 418.977 | -37.510 | -128.535 |
| Complex⁃1 | -265.140 | -363.046 | 538.899 | -38.367 | -127.654 |
| Complex⁃2 | -231.166 | -175.561 | 343.339 | -32.928 | -96.316 |
| Complex⁃3 | -194.263 | -239.492 | 362.502 | -24.811 | -96.106 |
Table 1 Predicted binding energies and individual energy components of the Dectin-1 complexes with CP and TH-bglc
| Gmx_mmpbsa | ΔEvdW/(kJ·mol-1) | ΔEelec/(kJ·mol-1) | ΔGpolar/(kJ·mol-1) | ΔGnonpolar/(kJ·mol-1) | ΔGbind/(kJ·mol-1) |
|---|---|---|---|---|---|
| TH⁃bglc | -231.681 | -278.322 | 418.977 | -37.510 | -128.535 |
| Complex⁃1 | -265.140 | -363.046 | 538.899 | -38.367 | -127.654 |
| Complex⁃2 | -231.166 | -175.561 | 343.339 | -32.928 | -96.316 |
| Complex⁃3 | -194.263 | -239.492 | 362.502 | -24.811 | -96.106 |
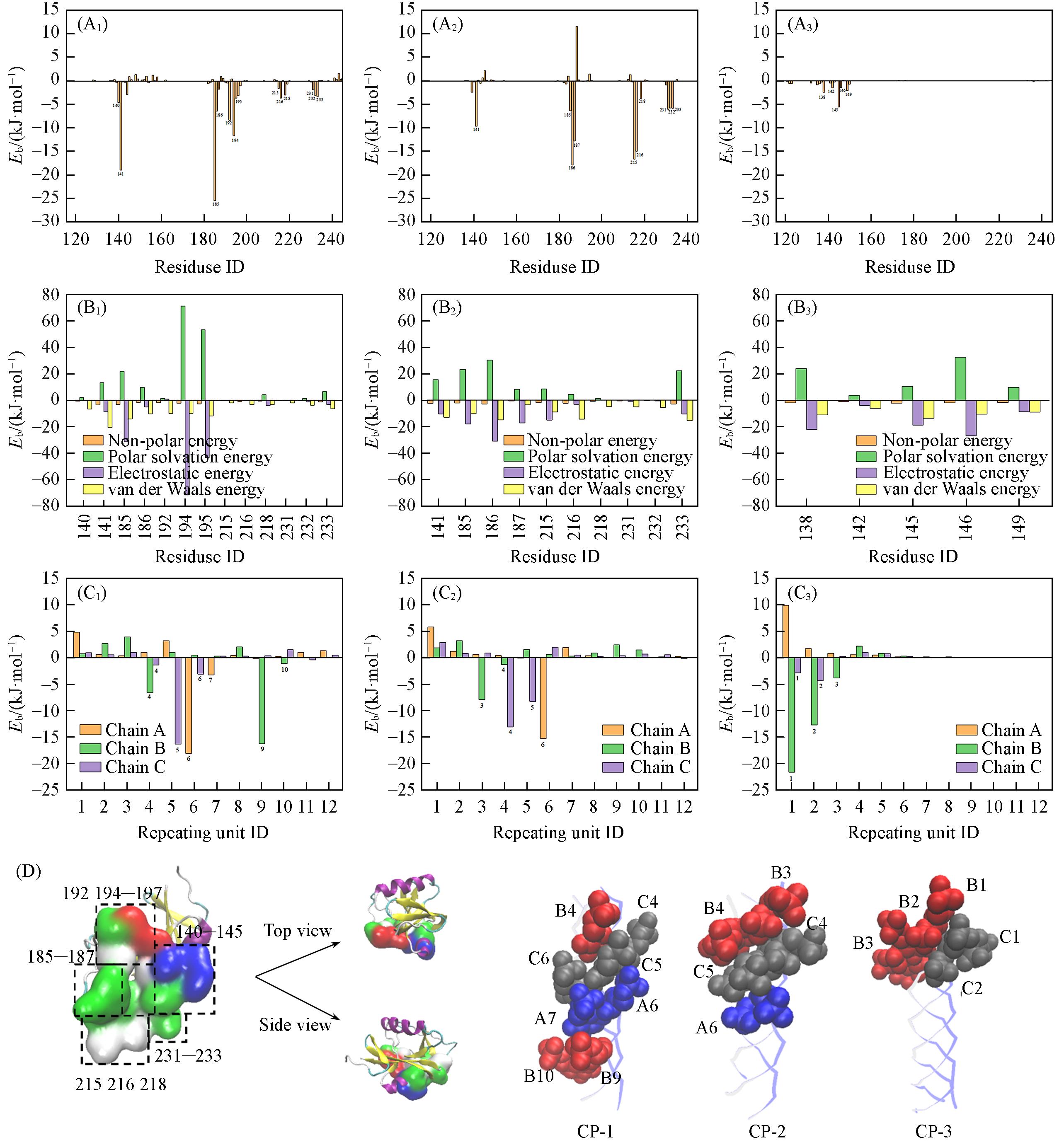
Fig.3 Residue binding energy analysis(A1—A3) Contributions of key amino acid residues in Dectin-1 to the total binding energy of the Dectin-1-CP-1(A1), Dectin-1-CP-2(A2), Dectin-1-CP-3(A3) complexes; (B1—B3) contributions of key amino acid residues in Dectin-1 to individual energy components(van der Waals, electrostatic, polar solvation, and non-polar solvation) in the Dectin-1-CP-1(B1), Dectin-1-CP-2(B2), Dectin-1-CP-3(B3) complexes; (C1—C3) energy decomposition profiles of the three chains(Chain A, Chain B and Chain C) in the Dectin-1-CP-1(C1), Dectin-1-CP-2(C2), Dectin-1-CP-3(C3) complexes(each panel shows the contributions of the three chains within one CP model); (D) schematic illustration of the recognition site region of Dectin-1 and the epitope recognition patterns of TH-CP samples with different substitutions patterns; the left panel shows the key residues forming the recognition region of Dectin-1 (top and side views); the right panels represent CP-1, CP-2, and CP-3 from left to right; the three chains of the triple helix are shown in different colors, highlighting the glucose units involved in epitope recognition from each individual chain.
| Dectin⁃1 residue | TH⁃CP chemical group* | Occupancy(%) | Dectin⁃1 residue | TH⁃CP chemical group* | Occupancy(%) |
|---|---|---|---|---|---|
| Tyr141 | C5PO63 | 14.9 | Asn185(O) | B4HPO61 | 30.3 |
| Lys144(HZ1) | A6PO63 | 82.1 | Glu194(O) | B9HPO61 | 54.7 |
| Lys144(HZ1) | B9PO63 | 37.8 | Glu194(OE1) | B9HPO62 | 16.9 |
| Arg145 | C4PO63 | 44.3 | Asp195(O) | B10HPO62 | 47.3 |
| Asn185(HD21) | B4O4 | 86.1 | Asp195(O) | A6HO4 | 33.3 |
| Asn185(HD21) | C5O6 | 79.6 | Asp195(O) | B10HPO61 | 24.4 |
| Asn185(O) | B4HPO62 | 51.2 |
Table 2 Hydrogen bond occupancies(>10%) between Dectin-1 and the O6 substitution CP-1 complex
| Dectin⁃1 residue | TH⁃CP chemical group* | Occupancy(%) | Dectin⁃1 residue | TH⁃CP chemical group* | Occupancy(%) |
|---|---|---|---|---|---|
| Tyr141 | C5PO63 | 14.9 | Asn185(O) | B4HPO61 | 30.3 |
| Lys144(HZ1) | A6PO63 | 82.1 | Glu194(O) | B9HPO61 | 54.7 |
| Lys144(HZ1) | B9PO63 | 37.8 | Glu194(OE1) | B9HPO62 | 16.9 |
| Arg145 | C4PO63 | 44.3 | Asp195(O) | B10HPO62 | 47.3 |
| Asn185(HD21) | B4O4 | 86.1 | Asp195(O) | A6HO4 | 33.3 |
| Asn185(HD21) | C5O6 | 79.6 | Asp195(O) | B10HPO61 | 24.4 |
| Asn185(O) | B4HPO62 | 51.2 |
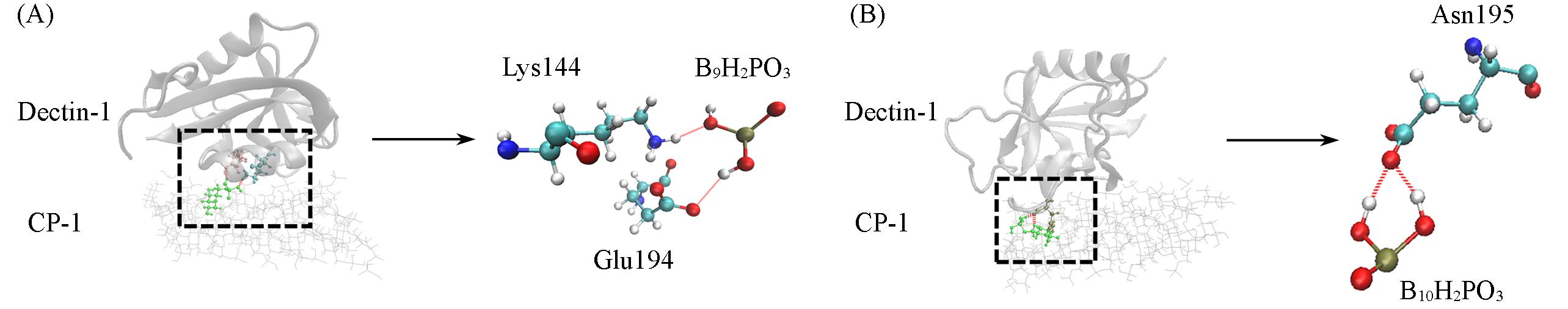
Fig.4 Schematic representation of the major hydrogen bonds formed between Dectin⁃1 and CP⁃1The portions of two characteristic hydrogen-bonding networks. Hydrogen bonds between Dectin-1 and CP-1 are indicated by red dashed lines. (A) Hydrogen-bonding network involving Lys144 and Glu194 residues of Dectin-1 with CP-1; (B) hydrogen-bonding network involving the Asn195 residue of Dectin-1 with CP-1. Hydrogen bonds are indicated by dashed lines.
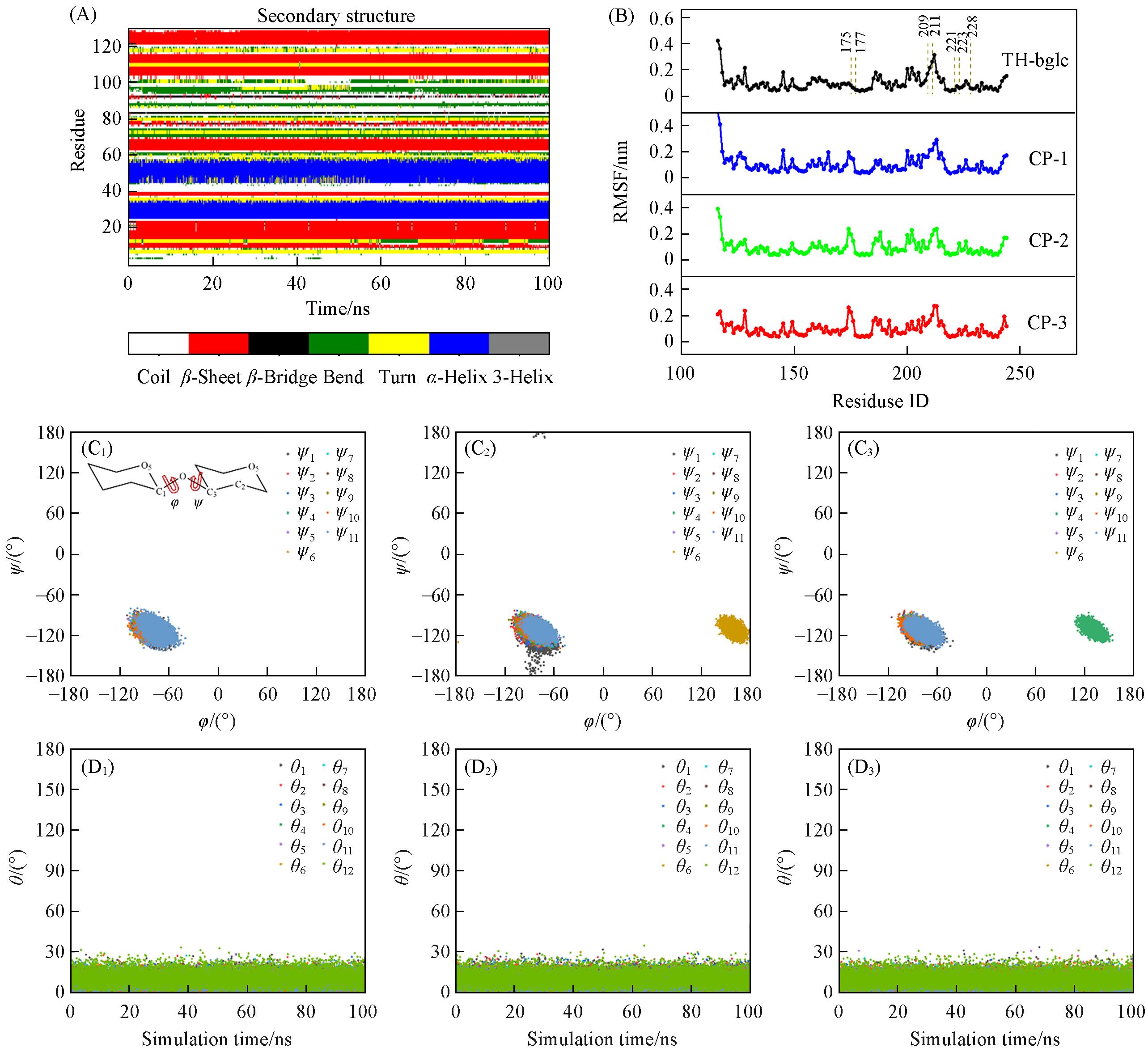
Fig.5 Structural analysis of CP⁃1 and its Dectin⁃1(A) Secondary structure(DSSP) of the Dectin-1 protein under O6 substitution; (B) root mean square fluctuation(RMSF) profiles of Dectin-1 residues in four systems: TH-bglc, CP-1, CP-2, and CP-3; residues 175—177, 209—211, and 221, 223, and 228 are highlighted by dashed lines; (C1—C3) distributions of glycosidic bond dihedral angles(φ and ψ) for each chain in the triple-helical conformation of CP-1; the panels correspond to Chain A(C1), Chain B(C2), and Chain C(C3), respectively, ψ1—ψ11 denote the glycosidic dihedral angles between adjacent glucose units along each chain; (D1—D3) fluctuations of the θ ring puckering parameter for each chain in CP-1, the panels correspond to Chain A(D1), Chain B(D2), and Chain C(D3), respectively, θ1—θ12 represent the ring puckering parameters of individual glucose residues, calculated based on the O5-C1-C2-C3-C4-C5 atom sequence.
| [62] | Noumi E., Snoussi M., Bouali N., Alshammari M. M., Altayb H. N., Afzal M., de Feo V., PLoS One, 2025, 20(7), e0324836—e0324837 |
| [63] | Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Acc. Chem. Res., 2000, 33(12), 889—897 |
| [64] | Homeyer N., Gohlke H., Mol. Inf., 2012, 31(2), 114—122 |
| [65] | Genheden S., Ryde U., Expert Opin. Drug Discovery, 2015, 10(5), 449—461 |
| [66] | Kumari R., Kumar R., Consortium O. S. D. D., Lynn A., J. Chem. Inf. Model., 2014, 54(7), 1951—1962 |
| [67] | Paissoni C., Spiliotopoulos D., Musco G., Spitaleri A., Comput. Phys. Commun., 2014, 185(11), 2920—2929 |
| [68] | Xie C. M., Lu Y. Y., An L. J., Wang Z. H., Wang J., Li M. L., Chem. J. Chinese Universities, 2025, 46(12), 20250247 |
| 谢冲墨, 卢宇源, 安立佳, 王振华, 王健, 李明伦. 高等学校化学学报, 2025, 46(12), 20250247 | |
| [69] | Bai R., Li S. W., Chen Q., Sun Z. Y., Xu W. S., Chem. J. Chinese Universities, 2024, 45(6), 20240013 |
| 白蓉, 李尚伟, 陈全, 孙昭艳, 徐文生. 高等学校化学学报, 2024, 45(6), 20240013 | |
| [70] | Guo X. H., Zhou Y. Z., Xie D. Q., Chem. Res. Chinese Universities, 2025, 41(5), 1076—1083 |
| [71] | Sun H. Y. J., Li X. H., Zeng X. L., Liu J., Rakmatullin A., Lou C. J., Tang M. X., Fernández⁃Carrión A. J., Kuang X. J., Chem. Res. Chinese Universities, 2025, 41(2), 296—304 |
| [72] | Zhang J. J., Lü L. N., Zhu H. R., Zhang Y., Xu X. D., Long L. X., Fu W., Chem. Res. Chinese Universities, 2024, 40(6), 1201—1211 |
| [73] | Nagae M., Yamaguchi Y., Int. J. Mol. Sci., 2014, 15(3), 3768—3783 |
| [74] | Baker E., International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules, Wiley, Hoboken, 2006, 546—552 |
| [1] | Caseiro C., Dias J. N. R., de Andrade Fontes C. M. G., Bule P., Int. J. Mol. Sci., 2022, 23(6), 3156—3157 |
| [2] | Saito H., Misaki A., Harada T., Agric. Biol. Chem., 1968, 32(10), 1261—1269 |
| [3] | Legentil L., Paris F., Ballet C., Trouvelot S., Daire X., Vetvicka V., Ferrières V., Molecules, 2015, 20(6), 9745—9766 |
| [4] | Manabe N., Yamaguchi Y., Int. J. Mol. Sci., 2021, 22(4), 1578—1579 |
| [5] | Aghaei M., Khademi R., Far M. A. J., Bahreiny S. S., Mahdizade A. H., Amirrajab N., Curr. Res. Transl. Med., 2024, 72(4), 103460— 103461 |
| [6] | Taylor P. R., Tsoni S. V., Willment J. A., Dennehy K. M., Rosas M., Findon H., Haynes K., Steele C., Botto M., Gordon S., Nat. Immunol., 2007, 8(1), 31—38 |
| [7] | Mansour M. K., Tam J. M., Khan N. S., Seward M., Davids P. J., Puranam S., Sokolovska A., Sykes D. B., Dagher Z., Becker C., J. Biol. Chem., 2013, 288(22), 16043—16054 |
| [8] | Tsoni S. V., Brown G. D., Ann. N. Y. Acad. Sci., 2008, 1143(1), 45—60 |
| [9] | Kimberg M., Brown G. D., Med. Mycol. Case Rep., 2008, 46(7), 631—636 |
| [10] | Marakalala M. J., Kerrigan A. M., Brown G. D., Mamm. Genome, 2011, 22(1), 55—65 |
| [11] | Cai Z., Zhang H., Carbohydr. Polym., 2021, 272, 118456—118457 |
| [12] | Zhang R. R., Edgar K. J., Biomacromolecules, 2014, 15(4), 1079—1096 |
| [13] | Guo X. Y., Kang J., Xu Z. Y., Guo Q. B., Zhang L. F., Ning H. F., Cui S. W., Carbohydr. Polym., 2021, 262, 117962—117963 |
| [14] | Meng Y., Lyu F. Z., Xu X. J., Zhang L. N., Biomacromolecules, 2020, 21(5), 1653—1677 |
| [15] | Feng X., Li F., Ding M. M., Zhang R., Shi T. F., Lu Y. Y., Jiang W., Carbohydr. Polym., 2022, 286, 119276—119277 |
| [16] | Liu H., Li Y., Gao J., Shi A., Liu L., Hu H., Putri N., Yu H., Fan W., Wang Q., Int. J. Biol. Macromol., 2016, 84, 394—401 |
| [17] | Šandula J., Kogan G., Kačuráková M., Machová E., Carbohydr. Polym., 1999, 38(3), 247—253 |
| [18] | Zekovic D. B., Kwiatkowski S., Vrvic M. M., Jakovljevic D., Moran C. A., Crit. Rev. Biotechnol., 2005, 25(4), 205—230 |
| [19] | Giese E. C., Covizzi L. G., Dekker R. F., Monteiro N. K., Da Silva M. D. L. C., Barbosa A. M., Process Biochem., 2006, 41(6), 1265—1271 |
| [20] | Li J., Zhu L., Zheng Z. Y., Zhan X. B., Lin C. C., Zong Y., Li W. J., Appl. Microbiol. Biotechnol., 2013, 97(19), 8495—8503 |
| [21] | Wang D., Kim D. H., Yoon J. J., Kim K. H., Process Biochem., 2017, 52, 233—237 |
| [22] | Chen X., Yu C., Wang J. H., Wu Y. C., Ma Y., Li H. J., Colloids Surf., A, 2023, 674, 131893—131894 |
| [23] | Mei X. Y., Tang Q. L., Huang G. L., Long R., Huang H. L., Food Chem., 2020, 309, 125791—125792 |
| [24] | Suflet D. M., Nicolescu A., Popescu I., Chitanu G. C., Carbohydr. Polym., 2011, 84(3), 1176—1181 |
| [25] | Xia S., Zhai Y., Wang X., Fan Q., Dong X., Chen M., Han T., Int. J. Biol. Macromol., 2021, 184, 946—954 |
| [26] | Shetty M. P., Tambe P., Rana K., Kulkarni S. D., Chaudhari P., Bharati S., Cell Biochem. Biophys., 2026, 84, 581—598 |
| [27] | Huang Q., Zhang L., Carbohydr. Polym., 2011, 83(3), 1363—1369 |
| [28] | Chen X. Y., Xu X. J., Zhang L. N., Zeng F. B., Carbohydr. Polym., 2009, 78(3), 581—587 |
| [29] | Chen F. M., Sun T., Song H. Z., J. Agric. Food Chem., 2026, 74(1), 40—58 |
| [30] | Pinho S. S., Alves I., Gaifem J., Rabinovich G. A., Cell. Mol. Immunol., 2023, 20(10), 1101—1113 |
| [31] | Han B., Baruah K., Cox E., Vanrompay D., Bossier P., Biophys. Rev. Lett., 2020, 11, 658—659 |
| [32] | Jiang S., Niu S., Yao W., Li Z. J., Li Q., Carbohydr. Res., 2016, 429, 148—154 |
| [33] | Zhang S., Chen K. Y., Zou X., Commun. Inf. Syst., 2021, 21(1), 147—148 |
| [34] | Mattox D. E., Bailey⁃Kellogg C., PLoS Comput. Biol., 2021, 17(10), 1009470—1009471 |
| [35] | Lei X. T., Jin Y. Q., Meng X. Y., Chem. J. Chinese Universities, 2021, 42(8), 2550—2557 |
| 雷晓彤, 金怡卿, 孟烜宇. 高等学校化学学报, 2021, 42(8), 2550—2557 | |
| [36] | Feng X., Li F., Ding M. M., Zhang R., Shi T. F., Jiang W., Carbohydr. Polym., 2021, 261, 117844—117845 |
| [37] | Feng X., Li F., Ding M. M., Zhang R., Shi T. F., Carbohydr. Polym., 2020, 250, 116906—116907 |
| [38] | Gao Y. F., Feng X., Zhang R., Xiao J., Huang Q. R., Li J. W., Shi T. F., Int. J. Biol. Macromol., 2024, 282, 137119—137120 |
| [39] | Hansen P. I., Spraul M., Dvortsak P., Larsen F. H., Blennow A., Motawia M. S., Engelsen S. B., Biopolymers, 2009, 91(3), 179—193 |
| [40] | Yan Y., Tao H., He J., Huang S. Y., Nat. Protoc., 2020, 15(5), 1829—1852 |
| [41] | Remmert M., Biegert A., Hauser A., Söding J., Nat. Methods, 2012, 9(2), 173—175 |
| [42] | Humphrey W., Dalke A., Schulten K., J. Mol. Graphics, 1996, 14(1), 33—38 |
| [43] | Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., SoftwareX, 2015, 1, 19—25 |
| [44] | Páll S., Zhmurov A., Bauer P., Abraham M., Lundborg M., Gray A., Hess B., Lindahl E., J. Chem. Phys., 2020, 153(13), 5728—5740 |
| [45] | Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., MacKerell A. D. Jr., Biophys. J., 2017, 112(3), 71—73 |
| [46] | Lee J., Cheng X., Jo S., MacKerell A. D., Klauda J. B., Im W., Biophys. J., 2016, 110(3), 405—413 |
| [47] | Liao S. W., Liu Y. C., Shi Z. N., Zhao D. H., Wei Y. Y., Li L. B., Chem. J. Chinese Universities, 2023, 44(10), 20230155 |
| 廖首维, 刘炎昌, 石泽南, 赵道辉, 魏嫣莹, 李理波. 高等学校化学学报, 2023, 44(10), 20230155 | |
| [48] | Jorgensen W. L., Tirado⁃Rives J., PNAS, 2005, 102(19), 6665—6670 |
| [49] | Su L. L., Shao X. G., Cai W. S., Chem. J. Chinese Universities, 2023, 44(4), 20220745 |
| 粟李醴, 邵学广, 蔡文生. 高等学校化学学报, 2022, 44(4), 20220745 | |
| [50] | Berendsen H. J., Postma J. V., van Gunsteren W. F., DiNola A., Haak J. R., J. Chem. Phys., 1984, 81(8), 3684—3690 |
| [51] | Hess B., Bekker H., Berendsen H. J., Fraaije J. G., J. Comput. Chem., 1997, 18(12), 1463—1472 |
| [52] | Darden T., York D., Pedersen L., J. Chem. Phys., 1993, 98, 10089—10090 |
| [53] | Kato K., Nakayoshi T., Kurimoto E., Oda A., Chem. Phys. Lett., 2021, 781, 139022—139023 |
| [54] | Kabsch W., Sander C., Biopolymers, 1983, 22(12), 2577—2637 |
| [55] | Henzler⁃Wildman K. A., Thai V., Lei M., Ott M., Wolf⁃Watz M., Fenn T., Pozharski E., Wilson M. A., Petsko G. A., Karplus M., 2007, 450(7171), 838—844 |
| [56] | Hollingsworth S. A., Dror R. O., Neuron, 2018, 99(6), 1129—1143 |
| [57] | Martínez L., PLoS One, 2015, 10(3), e0119264—e0119265 |
| [58] | Gorelov S., Titov A., Tolicheva O., Konevega A., Shvetsov A., J. Chem. Inf. Model., 2024, 64(9), 3593—3598 |
| [59] | Perez S., Makshakova O., Chem. Rev., 2022, 122(20), 15914—15970 |
| [60] | Lutsyk V., Wolski P., Plazinski W., J. Chem. Theory Comput., 2024, 20(14), 6350—6368 |
| [61] | Li J., gmxtools, 2022 |
| [1] | 刘莉莉, 杨佩, 田益嘉, 黄逸瑜, 张正东, 闫伟, 张原原, 史林兴. 高密度磷酸化纳米洋葱碳/磺化聚芳醚砜复合膜的制备与性能[J]. 高等学校化学学报, 2026, 47(2): 20250256. |
| [2] | 韩玉贵, 刘长龙, 赵鹏, 郑雯雯, 刘岳鹏, 李轶. 热水化学驱体系与稠油组分间相互作用及其理论模拟研究[J]. 高等学校化学学报, 2024, 45(6): 20230456. |
| [3] | 房意, 李英杰, 张友浩, 任宇, 韩奎华, 赵建立. 基于分子动力学的热化学储能过程中CaO/Ca(OH)2分子扩散机制研究[J]. 高等学校化学学报, 2024, 45(5): 20240052. |
| [4] | 富忠恒, 陈翔, 姚楠, 余乐耕, 沈馨, 张睿, 张强. 固态电解质锂离子输运机制研究进展[J]. 高等学校化学学报, 2023, 44(5): 20220703. |
| [5] | 沈琦, 陈海瑶, 高登辉, 赵熹, 那日松, 刘佳, 黄旭日. 天然产物法卡林二醇与人类GABAA受体相互作用的机制[J]. 高等学校化学学报, 2023, 44(2): 20220500. |
| [6] | 李吉辰, 蔡珊珊, 彭巨擘, 李宏飞, 段晓征. 电场下离子型聚合物复合囊泡结构变化的分子动力学模拟[J]. 高等学校化学学报, 2023, 44(2): 20220553. |
| [7] | 郝清海, 杨帆, 卿澈, 谭红革. 静电强度和反离子价态诱导的聚两性离子刷表面形貌[J]. 高等学校化学学报, 2023, 44(12): 20230279. |
| [8] | 周子豪, 王思皓, 黄玳川, 刘波, 甯红波. 正丙苯高温氧化机理的分子动力学模拟研究[J]. 高等学校化学学报, 2023, 44(11): 20230276. |
| [9] | 廖首维, 刘炎昌, 石泽南, 赵道辉, 魏嫣莹, 李理波. 水/石墨烯界面离子吸附的分子动力学模拟: 力场参数优化与吸附机制[J]. 高等学校化学学报, 2023, 44(10): 20230155. |
| [10] | 高志伟, 李军委, 史赛, 付强, 贾钧儒, 安海龙. 基于分子动力学模拟的TRPM8通道门控特性分析[J]. 高等学校化学学报, 2022, 43(6): 20220080. |
| [11] | 胡波, 朱昊辰. 双层氧化石墨烯纳米体系中受限水的介电常数[J]. 高等学校化学学报, 2022, 43(2): 20210614. |
| [12] | 雷晓彤, 金怡卿, 孟烜宇. 基于分子模拟方法预测PIP2在双孔钾通道TREK-1上结合位点的研究[J]. 高等学校化学学报, 2021, 42(8): 2550. |
| [13] | 李聪聪, 刘明皓, 韩佳睿, 朱镜璇, 韩葳葳, 李婉南. 基于分子动力学模拟的VmoLac非特异性底物催化活性的理论研究[J]. 高等学校化学学报, 2021, 42(8): 2518. |
| [14] | 曾永辉, 言天英. 质子水合结构的振动态密度分析[J]. 高等学校化学学报, 2021, 42(6): 1855. |
| [15] | 齐人睿, 李明昊, 常浩, 付学奇, 高波, 韩葳葳, 韩璐, 李婉南. 基于拉伸分子动力学模拟的黄嘌呤氧化酶抑制剂解离途径的理论研究[J]. 高等学校化学学报, 2021, 42(3): 758. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||