

Chem. J. Chinese Universities ›› 2026, Vol. 47 ›› Issue (2): 20250250.doi: 10.7503/cjcu20250250
• Physical Chemistry • Previous Articles Next Articles
LI Haojing1, GE Changwei1, ZHONG Qidi2, YAN Hong1( ), SUN Guohui1
), SUN Guohui1
Received:2025-09-08
Online:2026-02-10
Published:2025-11-10
Contact:
YAN Hong
E-mail:hongyan@bjut.edu.cn
CLC Number:
TrendMD:
LI Haojing, GE Changwei, ZHONG Qidi, YAN Hong, SUN Guohui. Theoritical Studies on the Photophysical Properties of 1,4-Dihydropyridine Derivatives[J]. Chem. J. Chinese Universities, 2026, 47(2): 20250250.
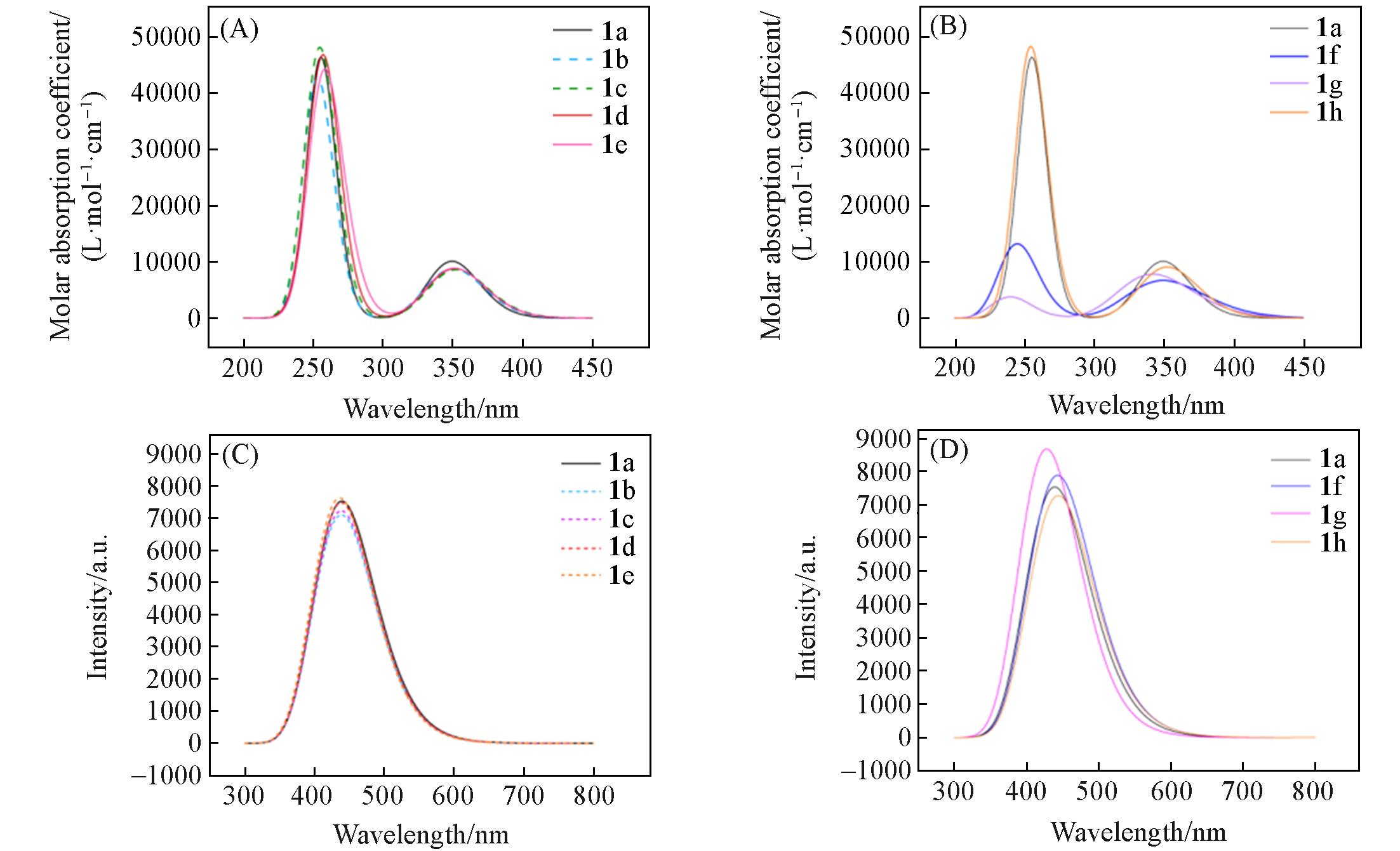
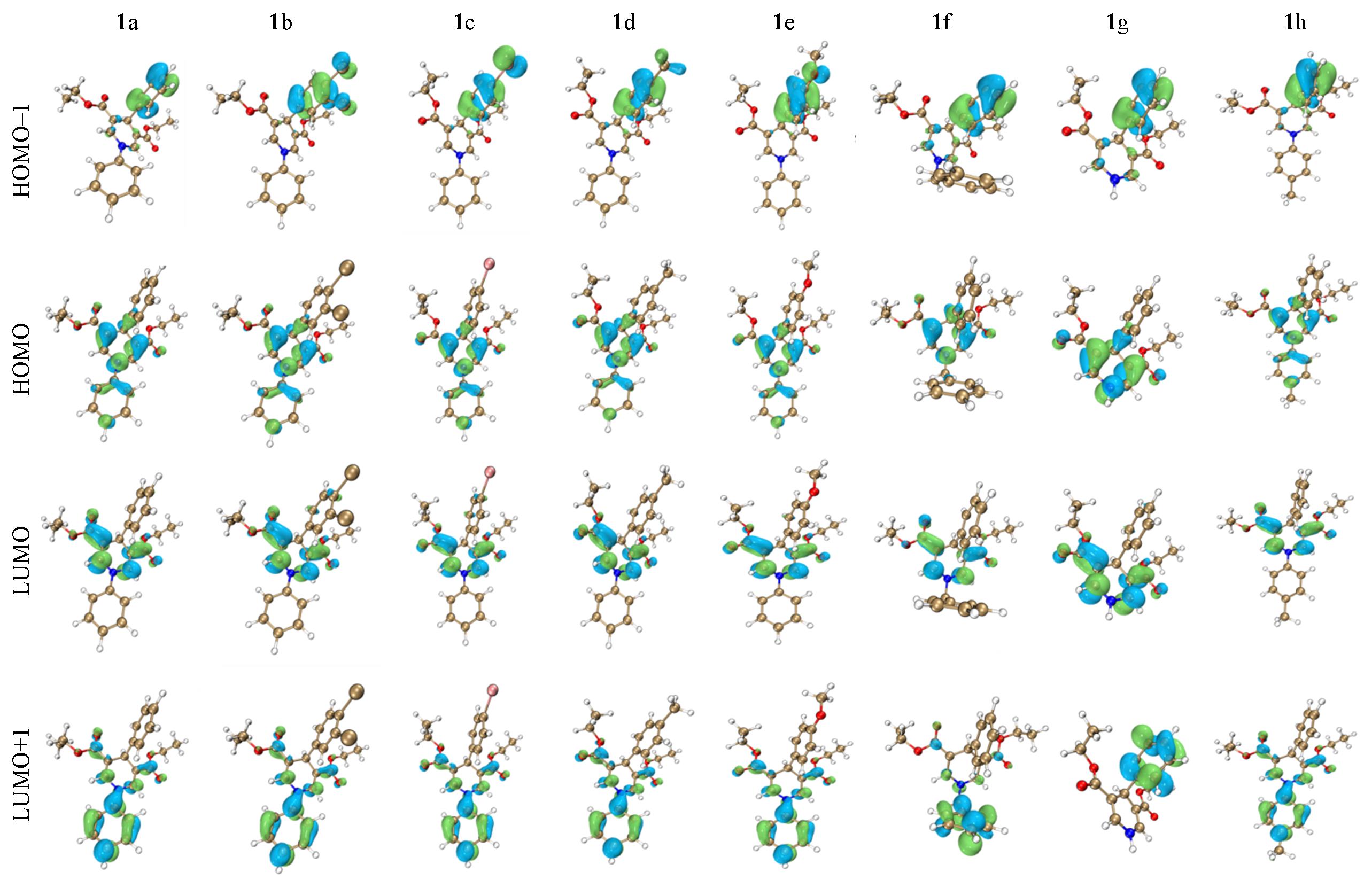
| Compound | EHOMO/eV | ELUMO/eV | ΔEHOMO-LUMO/eV |
|---|---|---|---|
| 1a | -7.1562 | -1.0553 | 6.1009 |
| 1b | -7.2416 | -1.1607 | 6.0809 |
| 1c | -7.1803 | -1.1181 | 6.0622 |
| 1d | -7.1155 | -1.0355 | 6.0799 |
| 1e | -7.1132 | -1.0296 | 6.0836 |
| 1f | -7.2550 | -1.0776 | 6.1774 |
| 1g | -7.2968 | -0.9861 | 6.3107 |
| 1h | -7.0889 | -1.0469 | 6.0420 |
Table 1 Molecular orbital energy levels of compounds 1a—1h
| Compound | EHOMO/eV | ELUMO/eV | ΔEHOMO-LUMO/eV |
|---|---|---|---|
| 1a | -7.1562 | -1.0553 | 6.1009 |
| 1b | -7.2416 | -1.1607 | 6.0809 |
| 1c | -7.1803 | -1.1181 | 6.0622 |
| 1d | -7.1155 | -1.0355 | 6.0799 |
| 1e | -7.1132 | -1.0296 | 6.0836 |
| 1f | -7.2550 | -1.0776 | 6.1774 |
| 1g | -7.2968 | -0.9861 | 6.3107 |
| 1h | -7.0889 | -1.0469 | 6.0420 |
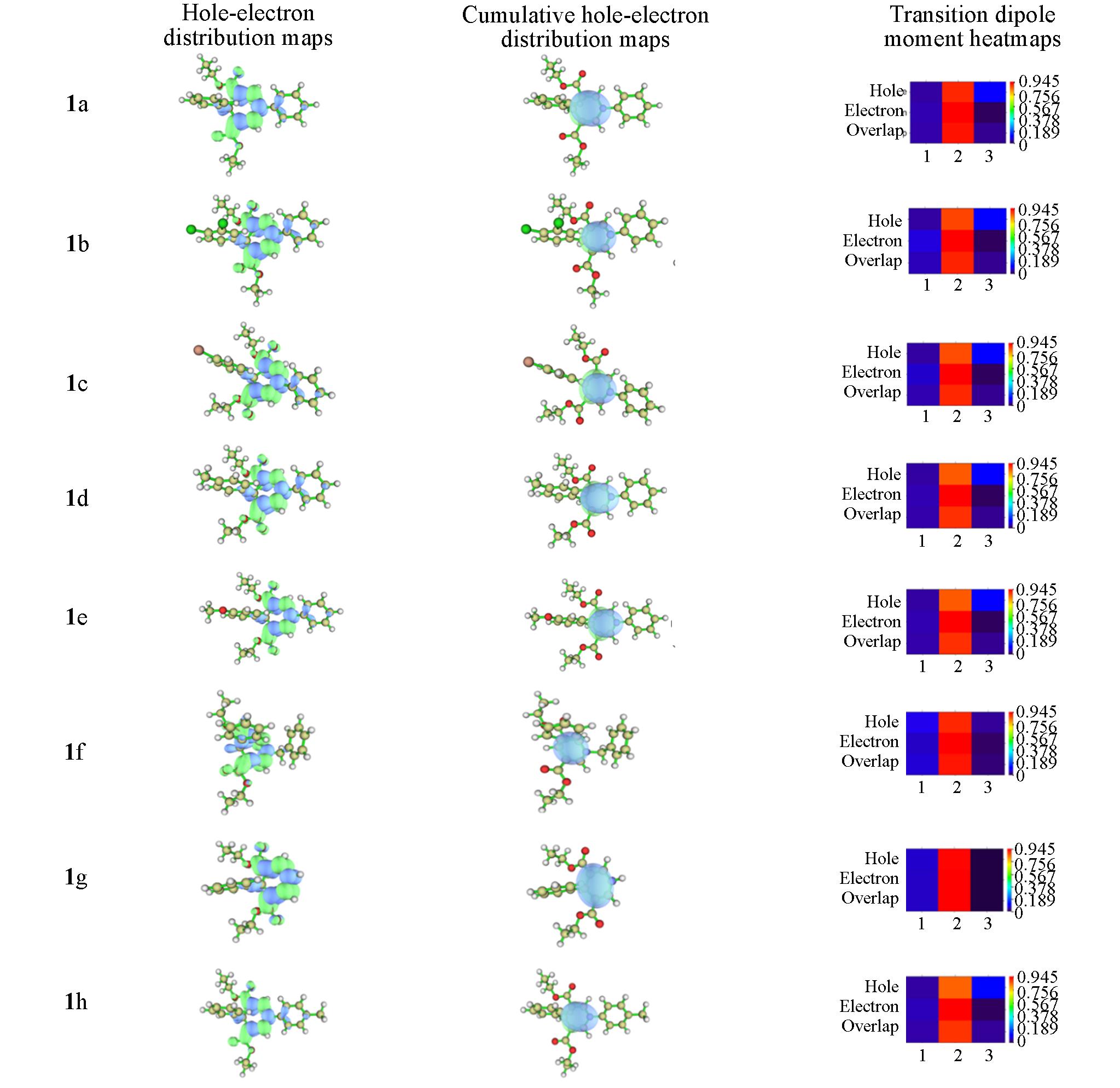
| Compound | Excitation index | Intrafragment rearrangement | ||||
|---|---|---|---|---|---|---|
| Overlap ratio | D | t | Fragment 1 | Fragment 2 | Fragment 3 | |
| 1a | 0.66078 | 0.0595 | -0.1065 | 0.00227 | 0.79881 | 0.00129 |
| 1b | 0.65760 | 0.0649 | -0.0950 | 0.00283 | 0.78474 | 0.00130 |
| 1c | 0.65810 | 0.0620 | -0.0945 | 0.00257 | 0.79052 | 0.00116 |
| 1d | 0.66031 | 0.0577 | -0.1051 | 0.00210 | 0.80076 | 0.00130 |
| 1e | 0.66005 | 0.0586 | -0.1015 | 0.00191 | 0.80507 | 0.00128 |
| 1f | 0.68625 | 0.0157 | -0.1215 | 0.00459 | 0.81850 | 0.00063 |
| 1g | 0.69800 | 0.0054 | -0.0982 | 0.00382 | 0.87821 | 0 |
| 1h | 0.65878 | 0.0645 | -0.1064 | 0.00224 | 0.78854 | 0.00153 |
Table 2 Relevant excited indexes of compounds 1a—1h
| Compound | Excitation index | Intrafragment rearrangement | ||||
|---|---|---|---|---|---|---|
| Overlap ratio | D | t | Fragment 1 | Fragment 2 | Fragment 3 | |
| 1a | 0.66078 | 0.0595 | -0.1065 | 0.00227 | 0.79881 | 0.00129 |
| 1b | 0.65760 | 0.0649 | -0.0950 | 0.00283 | 0.78474 | 0.00130 |
| 1c | 0.65810 | 0.0620 | -0.0945 | 0.00257 | 0.79052 | 0.00116 |
| 1d | 0.66031 | 0.0577 | -0.1051 | 0.00210 | 0.80076 | 0.00130 |
| 1e | 0.66005 | 0.0586 | -0.1015 | 0.00191 | 0.80507 | 0.00128 |
| 1f | 0.68625 | 0.0157 | -0.1215 | 0.00459 | 0.81850 | 0.00063 |
| 1g | 0.69800 | 0.0054 | -0.0982 | 0.00382 | 0.87821 | 0 |
| 1h | 0.65878 | 0.0645 | -0.1064 | 0.00224 | 0.78854 | 0.00153 |
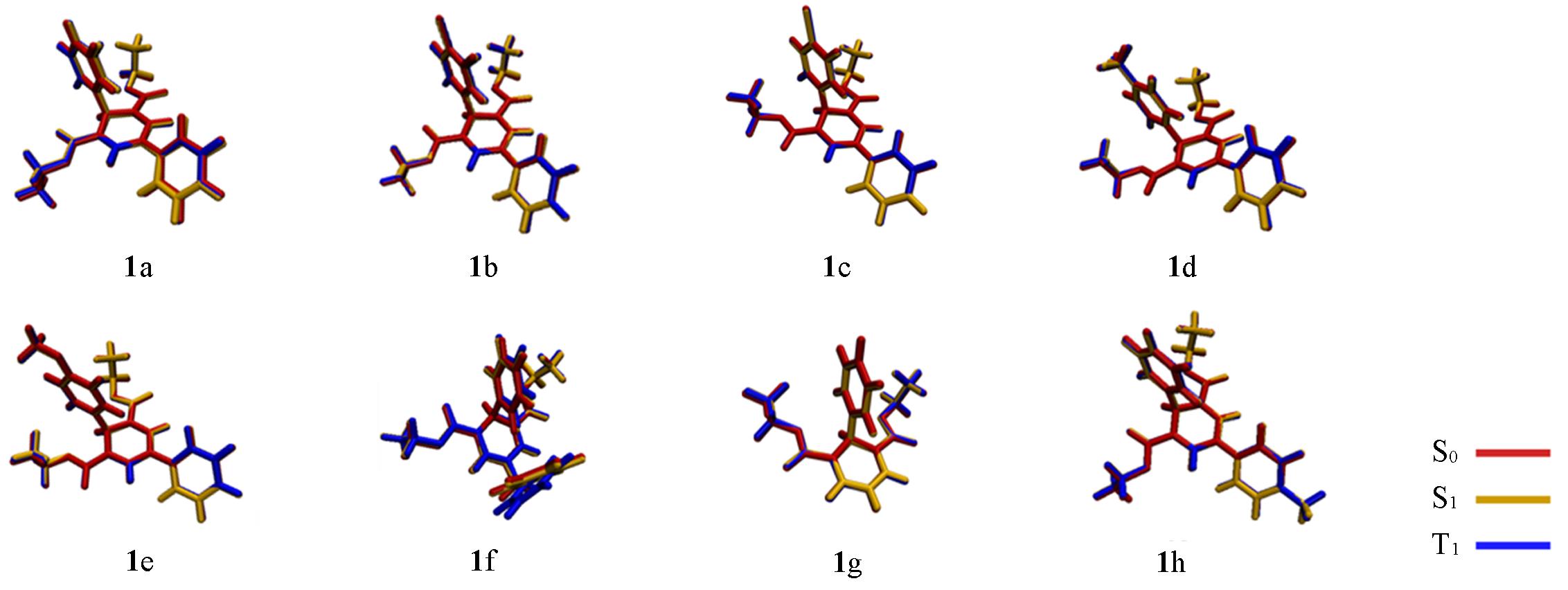
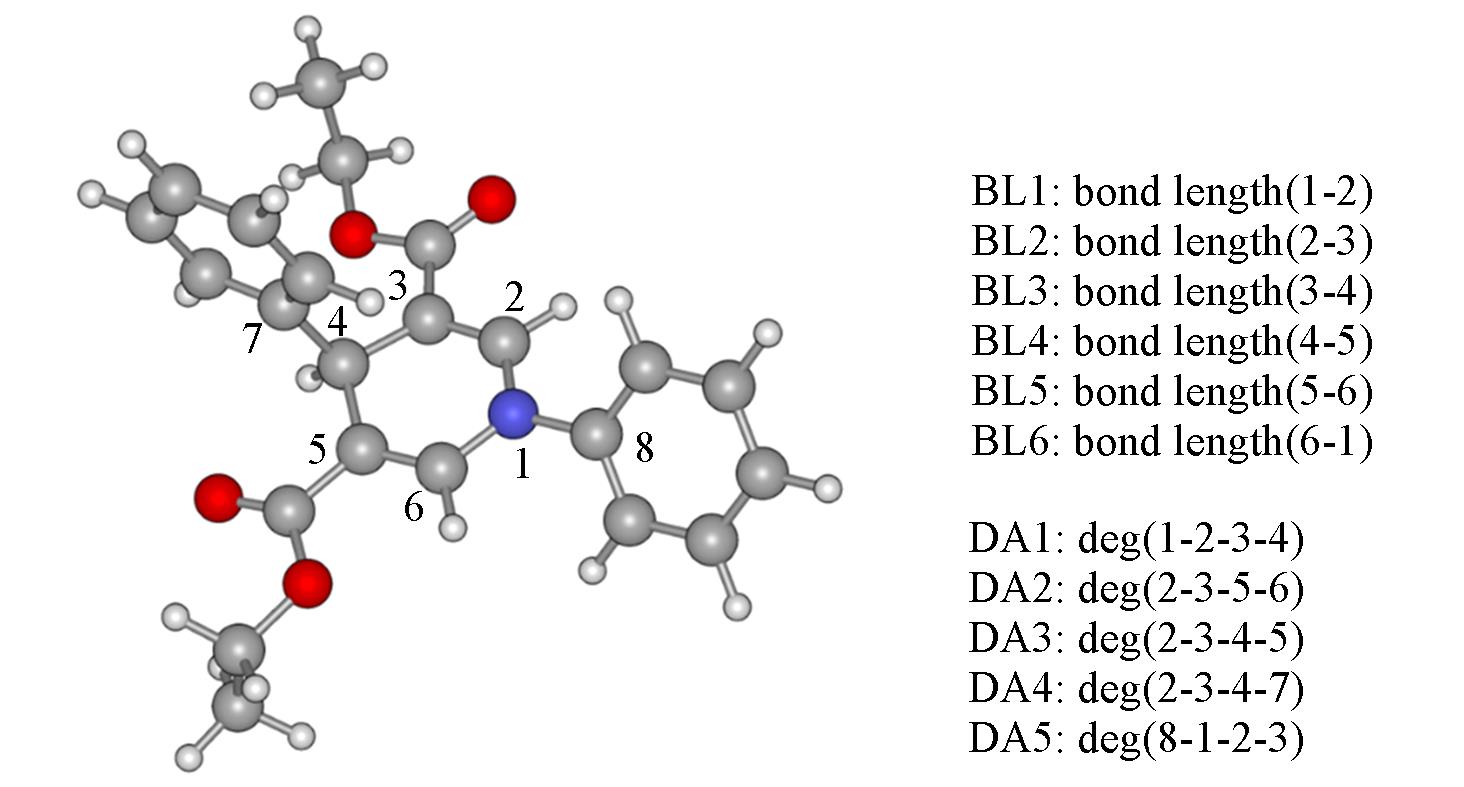
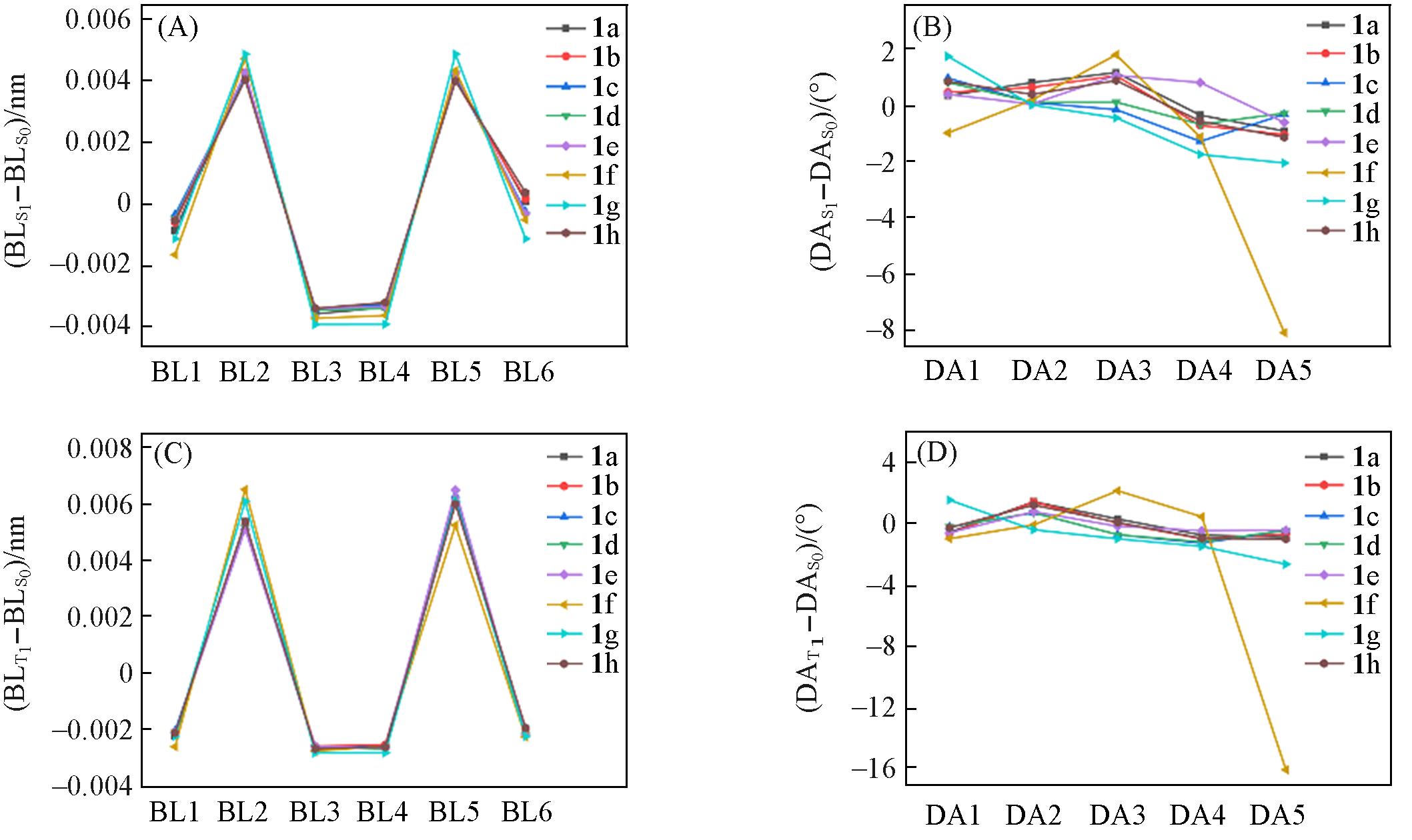
| [1] | Wang C., Lu Z., Org. Lett., 2017, 19(21), 5888—5891 |
| [2] | Zhang Y., Tanabe Y., Kuriyama S., Nishibayashi Y., J. Org. Chem., 2021, 86(18), 12577—12590 |
| [3] | Zhong Q., Fan Q., Yan H., Tetrahedron Lett., 2017, 58(13), 1292—1295 |
| [4] | Yan Z., Li J., Zheng Y., Miao Z., Chin. J. Chem. Educ., 2023, 44(18), 1—16 |
| [5] | Chaudhari S. R., Shirkhedkar A. A., Chin. J. Org. Chem., 2021, 51(3), 268—277 |
| [6] | Liu Y., Tan H., Yan H., Song X., Chem. Biol. Drug Des., 2013, 82(5), 567—578 |
| [7] | Manna D., Bhuyan R., Ghosh R., J. Mol. Model., 2018, 24(12), 340 |
| [8] | Ong S. T., Nam Y. W., Nasburg J. A., Ramanishka A., Ng X. R., Zhuang Z., Goay S. S. M., Nguyen H. M., Singh L., Singh V., Rivera A., Eyster M. E., Xu Y., Alper S. L., Wulff H., Zhang M., Chandy K. G., PNAS, 2025, 122(18), e2425494122 |
| [9] | Trivedi A. R., Dodiya D. K., Dholariya B. H., Kataria V. B., Bhuva V. R., Shah V. H., Bioorg. Med. Chem. Lett., 2011, 21(18), 5181—5183 |
| [10] | Valente S., Mellini P., Spallotta F., Carafa V., Nebbioso A., Polletta L., Carnevale I., Saladini S., Trisciuoglio D., Gabellini C., Tardugno M., Zwergel C., Cencioni C., Atlante S., Moniot S., Steegborn C., Budriesi R., Tafani M., Del Bufalo D., Altucci L., Gaetano C., Mai A., J. Med. Chem., 2016, 59(4), 1471—1491 |
| [11] | Zhu H., Chen S. J., Lin Y. X., Zhong Q. D., Sun W. J., Zhang X. J., Z. Kristallogr. New Cryst. Struct., 2019, 234(4), 793—794 |
| [12] | Chen X., Niu N., Li D., Zhang Z., Zhuang Z., Yan D., Li J., Zhao Z., Wang D., Tang B. Z., Adv. Funct. Mater., 2023, 33(8), 2211571 |
| [13] | Lian C., Zhang J., Mo F., Org. Chem. Front., 2024, 11(4), 1140—1149 |
| [14] | Fan Q., Tan H., Li P., Yan H., New J. Chem., 2018, 42(20), 16795—16805 |
| [15] | Zhu X., Li W., Yan H., Zhong R., J. Photochem. Photobiol. A: Chem., 2012, 241, 13—20 |
| [16] | Zhu X. H., Ni C. L., Song X. Q., Yan H., Zhong R. G., Chin. J. Org. Chem., 2010, 30(2), 276—281 |
| [17] | Sun W., Fan Q., Yan H., J. Photochem. Photobiol. A: Chem., 2018, 359, 33—39 |
| [18] | Tan H. B., Zhao Z. C., Ma Z. S., Yan H., Tetrahedron, 2018, 74(5), 529—534 |
| [19] | Wang S., Wang Y., Ge C., Sun R., Wang H., Yan H., J. Mol. Struct., 2023, 1273, 134316 |
| [20] | Zhong Q. D., Study on Photochemical Reactions and Mechanism of 1,4⁃Dihydropyridine Derivatives, Beijing University of Technology, Beijing, 2017 |
| 钟启迪. 1,4-二氢吡啶衍生物的光化学反应及反应机理研究, 北京: 北京工业大学, 2017 | |
| [21] | Sun R., Song X., Wang S., Zhang X., Yan H., Wang Y., Chin. Chem. Lett., 2023, 34(12), 108183 |
| [22] | Zhang X., Wei C., Zhang Y., Yan H., Li P., J. Mol. Struct., 2024, 1306, 137893 |
| [23] | Frisch M. J., Pople J. A., Binkley J. S., J. Chem. Phys., 1984, 80(7), 3265—3269 |
| [24] | Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem., 2011, 32(7), 1456—1465 |
| [25] | Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem., 1994, 98(45), 11623—11627 |
| [26] | Cammi R., Tomasi J., J. Chem. Phys., 1994, 100(10), 7495—7502 |
| [27] | Miertuš S., Scrocco E., Tomasi J., Chem. Phys., 1981, 55(1), 117—129 |
| [28] | Lu T., Chen F., J. Comput. Chem., 2012, 33(5), 580—592 |
| [29] | Lu T., J. Chem. Phys., 2024, 161(8), 082503 |
| [30] | Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 1996, 14(1), 33—38 |
| [31] | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams Y., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J., Gaussian 16, Rev. B.01, Wallingford CT, Gaussian Inc., 2016 |
| [32] | Fan Q., Li P., Yan H., J. Photochem. Photobiol. A: Chem., 2018, 358, 51—60 |
| [33] | He Y., Wang H., Ge C., Yan H., J. Mol. Struct., 2023, 1281, 135167 |
| [34] | Li P., Wang S., Tian N., Yan H., Wang J., Song X., Org. Biomol. Chem., 2021, 19(17), 3882—3892 |
| [35] | Costa R. A., Pitt P. O., Pinheiro M. L. B., Oliveira K. M. T., Salomé K. S., Barison A., Costa E. V., Spectrochim. Acta A: Mol. Biomol. Spectrosc., 2017, 174, 94—104 |
| [1] | CHENG Rou, SUN Fanqi, LU Xinhuan, GUO Haotian, ZHAN Junhui, HUANG Jia, YAN Shan, ZHOU Dan, XIA Qinghua. Sustainable Epoxidation of Cycloalkenes Using Air Catalyzed by Bimetallic CuCo-MOFs [J]. Chem. J. Chinese Universities, 2026, 47(2): 20250232. |
| [2] | XU Yiming, SHI Yiwei, WANG Xin, ZHU Zhihui, SONG Zhiguo, WANG Min. Green Synthesis of Mono- and Disubstituted Quinazolinones by Multi-site Synergistic Catalysis of Novel Three-dimensional Single-nuclear Zn(II) Complexes [J]. Chem. J. Chinese Universities, 2026, 47(2): 20250225. |
| [3] | KUANG Qin, ZHENG Lansun, ZHU Yaxian. Practice and Exploration on Curriculum Construction of Inorganic Chemistry Under the Chemistry "101 Plan" [J]. Chem. J. Chinese Universities, 2026, 47(2): 20260025. |
| [4] | LUO Shufang, ZHAO Yuanjin, WANG Shuo, ZHOU Runchaun, YANG Xia, HE Aihua. Preparation of Amine-capped Functionalized Trans-1,4-poly(butadiene-co-isoprene) Rubber and Coordination Chain Transfer Mechanism [J]. Chem. J. Chinese Universities, 2025, 46(8): 20250067. |
| [5] | WANG Zhiyuan, DONG Yi, QI Baohui, WEI Xueyang, ZHANG Jiahui, HUANG Qizhong, LI Jisheng, GAO Na, DI Shiying, HU Yufeng. Determination of Acidic Ionic Liquid H0 and the Effect of Salt Effect [J]. Chem. J. Chinese Universities, 2025, 46(7): 20240535. |
| [6] | LI Dan, HU Honghui, HOU Hongshuai, ZHANG Sheng, LIU Lijie, JING Mingjun, WU Tianjing. Sodium Storage Performance of Mixed-phase Sodium Titanate Tuned by Carbon Dots [J]. Chem. J. Chinese Universities, 2025, 46(6): 20240356. |
| [7] | XU Xingyu, XIE Xiaoming, QIU Ping. Ring-opening Mechanism of 2-Phenyl-3-amineazetidine to Form Thiazole or Oxazole [J]. Chem. J. Chinese Universities, 2025, 46(5): 20240547. |
| [8] | LUO Kui, LIN Jiaxi, LI Jianping. Development of a Glycosyl-imprinted Sensor and Rapid Detection of PD-L1 Positive Exosomes in Breast Cancer [J]. Chem. J. Chinese Universities, 2025, 46(5): 20240524. |
| [9] | HU Yuteng, SANG Lixia, DU Chunxu. Interfacial Performances of MoS2-H2O Depended on Plasmonic Metal and Its Localized Thermal Effect [J]. Chem. J. Chinese Universities, 2025, 46(5): 20240569. |
| [10] | YANG Siwei, HUANG Xuri. Theoretical Study of B, N Co-doped Fullerene C70 as Non-metal Electrocatalysts for Oxygen Reduction and Evolution [J]. Chem. J. Chinese Universities, 2025, 46(4): 20240490. |
| [11] | SHEN Yuhao, TIAN Zemin, LI Wei, JI Yixuan, YAN Yingwen. Theoretical Study of the Effect of Conformational Structures on the Secondary Oxidation Reactions of cis-1,3-Dimethylcyclohexane [J]. Chem. J. Chinese Universities, 2025, 46(3): 20240458. |
| [12] | LIU Yixuan, HU Huimin, FAN Xiaoqiang, YU Xuehua, KONG Lian, XIAO Xia, XIE Zean, ZHAO Zhen. Preparation of Pt/Mn-silicalite-1 Catalysts and Their Catalytic Performance for Propane Dehydrogenation [J]. Chem. J. Chinese Universities, 2025, 46(3): 20240460. |
| [13] | HUANG Zhiyao, LI Li, XU Huaqing, YANG Yifan, WEI Yaoyao, LIU Guokui, XIA Qiying. First-principles Study on the Catalysis of OER/ORR by N-doped Graphene with Defects [J]. Chem. J. Chinese Universities, 2025, 46(2): 20240430. |
| [14] | LIU Meng, XU Yi, YANG Fan, ZHOU Quan, REN Jing, REN Ruipeng, LYU Yongkang. Synthesis of COF-LZU1 in Acetate Buffer and Immobilized Enzyme Study [J]. Chem. J. Chinese Universities, 2025, 46(2): 20240368. |
| [15] | REN Zhihao, ZUO Teng, ZHANG Weiyun, YU Dahai, FANG Xuexun. Expression and Purification of Recombinant Amuc_0119 Protein Encoded by the Gut Commensal Bacterium Akkermansia Muciniphila [J]. Chem. J. Chinese Universities, 2025, 46(12): 20250283. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||