

Chem. J. Chinese Universities ›› 2015, Vol. 36 ›› Issue (11): 2292.doi: 10.7503/cjcu20150689
• Physical Chemistry • Previous Articles Next Articles
HU Xue, SUN Mingjun, ZHANG Xiangfei, CAO Zexing*( )
)
Received:2015-09-07
Online:2015-11-10
Published:2015-10-26
Contact:
CAO Zexing
E-mail:zxcao@xmu.edu.cn
Supported by:CLC Number:
TrendMD:
HU Xue, SUN Mingjun, ZHANG Xiangfei, CAO Zexing. Theoretical Study of Electronic Spectra of Lignite Structural Units†[J]. Chem. J. Chinese Universities, 2015, 36(11): 2292.
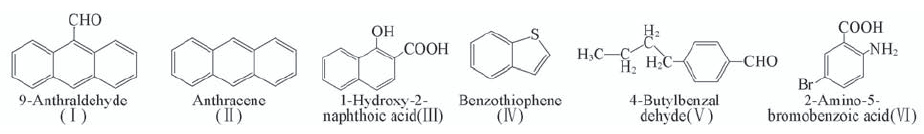
Fig.1 Six selected compounds for calibration of density functionals in calculation of electronic absorption spectra
| Compound | λmax/nm | λmax/nm(f), assignment*(contribution to the excited state) | |||||
|---|---|---|---|---|---|---|---|
| B3LYP | CAM-B3LYP | HSEH1PBE | ωB97X | M062X | ωB97XD | ||
| Ⅰ | 400 | 430(0.13), H→L(70%) | 380(0.18), H→L(70%) | 422(0.13), H→L(70%) | 359(0.21), H→L(70%) | 377(0.17), H→L(70%) | 379(0.19), H→L(70%) |
| Ⅱ | 375 | 386(0.13), H→L(70%) | 343(0.11), H→L(70%) | 380(0.08), H→L(70%) | 325(0.13), H→L(70%) | 341(0.11), H→L(70%) | 341(0.11), H→L(70%) |
| Ⅲ | 258 | 266(0.13), H→L(60%) | 250(0.08), H→L(50%) | 260(0.10), H→L(50%) | 245(0.07), H→L(47%) | 249(0.09), H→L(53%) | 251(0.08), H→L(53%) |
| Ⅳ | 340 | 333(0.13), H→L(68%) | 302(0.14), H→L(66%) | 302(0.14), H→L(66%) | 290(0.16), H→L(64%) | 298(0.15), H→L(66%) | 301(0.15), H→L(66%) |
| Ⅴ | 238 | 241(0.13), H→L(69%) | 226(0.42), H→L(68%) | 226(0.44), H→L(68%) | 221(0.39), H→L(67%) | 223(0.46), H→L(69%) | 226(0.42), H→L(68%) |
| Ⅵ | 324 | 329(0.13), H→L(70%) | 301(0.13), H→L(69%) | 322(0.11), H→L(70%) | 290(0.14), H→L(68%) | 298(0.13), H→L(69%) | 301(0.13), H→L(69%) |
Table 1 Predicted electronic absorptions(λmax/nm), oscillator strengths(f), and assignments for six compounds by different approaches
| Compound | λmax/nm | λmax/nm(f), assignment*(contribution to the excited state) | |||||
|---|---|---|---|---|---|---|---|
| B3LYP | CAM-B3LYP | HSEH1PBE | ωB97X | M062X | ωB97XD | ||
| Ⅰ | 400 | 430(0.13), H→L(70%) | 380(0.18), H→L(70%) | 422(0.13), H→L(70%) | 359(0.21), H→L(70%) | 377(0.17), H→L(70%) | 379(0.19), H→L(70%) |
| Ⅱ | 375 | 386(0.13), H→L(70%) | 343(0.11), H→L(70%) | 380(0.08), H→L(70%) | 325(0.13), H→L(70%) | 341(0.11), H→L(70%) | 341(0.11), H→L(70%) |
| Ⅲ | 258 | 266(0.13), H→L(60%) | 250(0.08), H→L(50%) | 260(0.10), H→L(50%) | 245(0.07), H→L(47%) | 249(0.09), H→L(53%) | 251(0.08), H→L(53%) |
| Ⅳ | 340 | 333(0.13), H→L(68%) | 302(0.14), H→L(66%) | 302(0.14), H→L(66%) | 290(0.16), H→L(64%) | 298(0.15), H→L(66%) | 301(0.15), H→L(66%) |
| Ⅴ | 238 | 241(0.13), H→L(69%) | 226(0.42), H→L(68%) | 226(0.44), H→L(68%) | 221(0.39), H→L(67%) | 223(0.46), H→L(69%) | 226(0.42), H→L(68%) |
| Ⅵ | 324 | 329(0.13), H→L(70%) | 301(0.13), H→L(69%) | 322(0.11), H→L(70%) | 290(0.14), H→L(68%) | 298(0.13), H→L(69%) | 301(0.13), H→L(69%) |
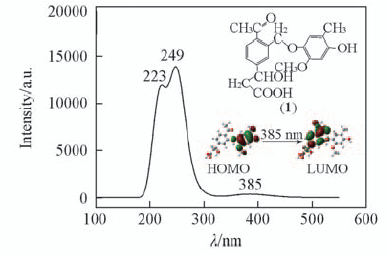
Fig.2 B3LYP-predicted absorption spectra and related frontier orbitals of the structural unit 1(inset)
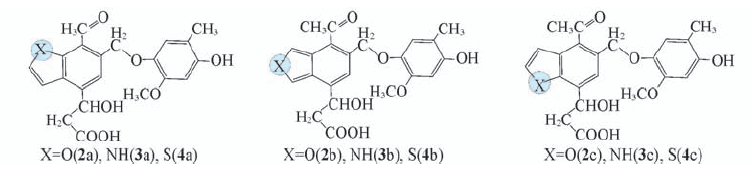
Fig.3 Structural units containing furan, pyrrole, or thiophene component for the lignite fragment
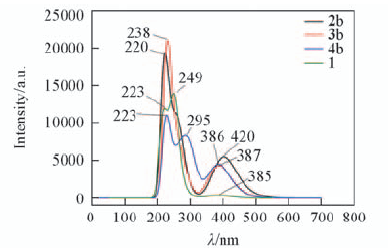
Fig.4 Predicted electronic spectra of the structural units 1, 2b, 3b and 4b
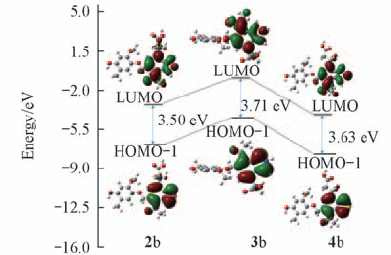
Fig.5 Selected frontier orbitals in structural units 2b, 3b and 4b
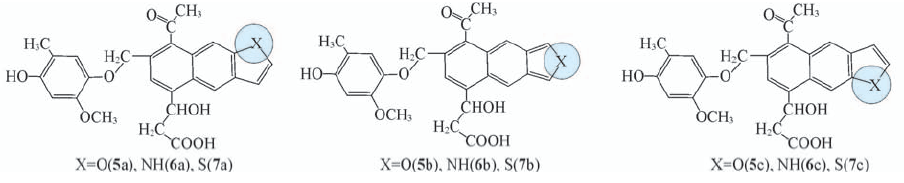
Fig.6 Selected tricyclic structural units for the lignite fragment
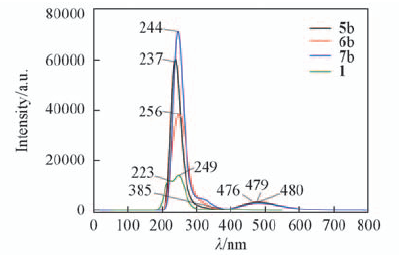
Fig.7 Predicted electronic spectra of structural units 1, 5b, 6b, and 7b
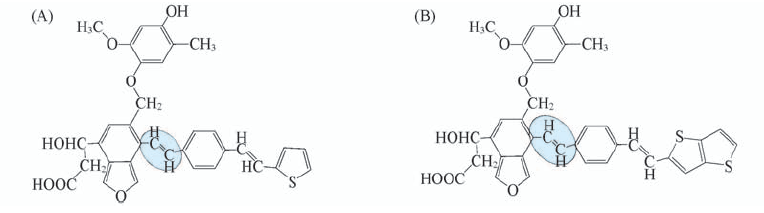
Fig.8 Structural units 8a(A) and 9a(B) for the lignite fragment
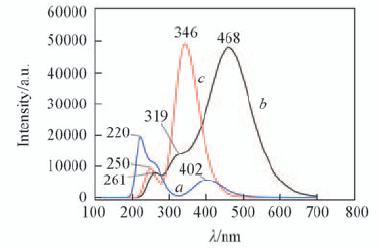
Fig.9 Predicted electronic spectra of structural units 2b(a), 8a(b) and 8b(c)
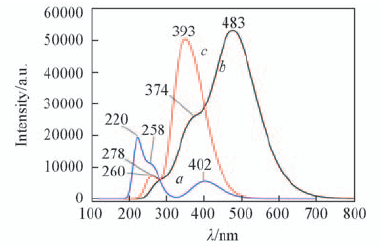
Fig.10 Predicted electronic spectra of structural units 2b(a), 9a(b) and 9b(c)
| [1] | Chen P., The Properties, Classification and Applications of Chinese Coal, Chemical Industry Press, Beijing, 2007, 24—25 |
| (陈鹏. 国煤炭性质、分类和利用, 北京: 化学工业出版社, 2007, 24—25) | |
| [2] | Wan Y. Z., Gao J. R., Xiao L., Tao X. X., Liu J. T., Coal Eng., 2010, 4, 75—79 |
| (万永周, 高俊荣, 肖雷, 陶秀祥, 刘炯天. 煤炭工程, 2010, 4, 75—79) | |
| [3] | Christian B., Fuels, 2004, 83(3), 267—276 |
| [4] | Shao J. J., Northwest Coal, 2009, 7(2), 17—22) |
| (邵俊杰. 西北煤炭, 2009, 7(2), 17—22) | |
| [5] | Guo J.J., Theoretical Studies of Structures and Properties of Coals, Xiamen Universities, Xiamen, 2013 |
| (郭娟娟. 煤结构与性质的理论研究, 厦门: 厦门大学, 2013) | |
| [6] | Wang J.R., Jin Z. X., Deng C. B., Quantum Chemistry Theory of Coal Spontaneous Combustion, Science Press, Beijing, 2007, 1—5 |
| (王继仁, 金智新, 邓存宝. 煤自燃量子化学理论, 北京: 科学出版社, 2007, 1—5) | |
| [7] | Li H., Zhang Y. T., Wang G. H., Zhou A. N., Appl. Chem. Ind., 2007, 36(4), 325—327 |
| (李慧, 张亚婷, 汪广恒, 周安宁. 应用化工, 2007, 36(4), 325—327) | |
| [8] | Jiang Y. F., Li K. S., Zhou A. N., Yang Z. Y., Coal Convers., 2004, 27(4), 83—86 |
| (姜玉凤, 李侃社, 周安宁, 杨致远. 煤炭转化, 2004, 27(4), 83—86) | |
| [9] | Zhang Y. T., Zhou A. N., J. Xi’an Univ. Sci. Tech., 2008, 2, 344—348 |
| (张亚婷, 周安宁. 西安科技大学学报, 2008, 2, 344—348 ) | |
| [10] | Carlo A., Denis J., Chem. Soc. Rev., 2013, 42, 845—846 |
| [11] | Jones R. N., Chem. Rev., 1947, 41(2), 353—371 |
| [12] | Muranaka A., Yasuike S., Liu C.Y., Kurita J., Kakusawa N., Tsuchiya T., Okuda M., Kobayashi N., Matsumoto Y., Yoshida K., Hashizumi D., Uchiyama M., J. Phys. Chem. A, 2009, 113(2), 464—473 |
| [13] | Mishra H., Maheshwary S., Tripathi H. B., Sathyamurthy N., J. Phys. Chem. A, 2005, 109, 2746—2754 |
| [14] | Karabacak M.,Cinar Z., Kurt M., Sudha S., Sundaraganesanc N., Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2012, 85, 179—189 |
| [15] | Karabacak M., Cinar M., Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2012, 86, 590—599 |
| [16] | Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., et al., Gaussian 09, Revision B. 01, Gaussian Inc., Wallingford CT, 2009 |
| [17] | Chen. C. G., Xian X. F., Coal Convers., 1998, 21(2), 7—12 |
| (陈昌国, 鲜学福. 煤炭转化, 1998, 21(2), 7—12) | |
| [18] | Wang S.Y., Study of Lignite Structure by Molecular Dynamics Simulation and Quantum Chemistry, Taiyuan University of Technology, Taiyuan, 2004 |
| (王三跃. 褐煤结构的分子动力学模拟及量子化学研究, 太原: 太原理工大学, 2004) | |
| [19] | Jia Y., The Experimental Analysis of Brown Coal Structure, Taiyuan University of Technology, Taiyuan, 2002 |
| (贾燕. 褐煤结构的实验分析, 太原: 太原理工大学, 2002) |
| [1] | HE Jinlu, LONG Run, FANG Weihai. A-site Cation Effects on Hot Carrier Relaxation in Perovskites by Nonadiabatic Molecular Dynamics Simulations † [J]. Chem. J. Chinese Universities, 2020, 41(3): 439. |
| [2] | TANG Haiyan, ZHAO Maoshuang, FENG Li, CAO Zexing. Theoretical Studies on the Taking off of Oxygen-containing Functional Groups in Lignite Model Compounds† [J]. Chem. J. Chinese Universities, 2014, 35(11): 2370. |
| [3] | ZHANG Jian-Po, JIN Li*, ZHANG Hong-Xing, BAI Fu-Quan. Structures and Spectroscopic Properties of Highly Efficient Luminescence Material Cationic [(C^N)2IrL]+ Complexes [J]. Chem. J. Chinese Universities, 2011, 32(12): 2885. |
| [4] | CHEN Jun-Jie, YI Yu-Ting, YU Miao, XUE Shao-Long, XIONG Qian-Qian, HE Xiao-Meng, ZHANG Lian-Ru*. Inhibitation and Binding of Rugulosin with N-Hsp90 [J]. Chem. J. Chinese Universities, 2011, 32(1): 88. |
| [5] | QIU Yi-Xiang, WANG Shu-Guang*. Time\|dependent Density Functional Theory Studies on the Electronic Spectra of Mo2 and W2 Quadruply Bonded Compounds [J]. Chem. J. Chinese Universities, 2011, 32(1): 161. |
| [6] | DONG She-Ying*, LI Jing, HUANG Ting-Lin. Microwave-induced Synthesis and Spectral Properties of New 4-Hydroxycoumarin Derivatives [J]. Chem. J. Chinese Universities, 2009, 30(8): 1516. |
| [7] | WANG Hui-Ping, BAI Fu-Quan, ZHENG Qing-Chuan, ZHAO Zeng-Xia, ZHANG Hong-Xing*. Theoretical Investigation on the Changes of Structures and Properties Caused by the Different Link Form in Bicarbazoles [J]. Chem. J. Chinese Universities, 2009, 30(12): 2434. |
| [8] | LI Ming-Xia, ZHOU Xin, ZHANG Hong-Xing*, FU Hong-Gang, SUN Chia-Chung. Theoretical Studies of Electronic Structures and Spectroscopic Properties of [M(N)X2]-(M=Ru, Os; X=S2C6H4, mnt) [J]. Chem. J. Chinese Universities, 2009, 30(11): 2284. |
| [9] | XIA Shu-Wei*, SHA Peng-Yan, YU Liang-Min, FAN Yu-Hua, BI Cai-Feng, YANG Li-Rong. TD-DFT Study on Electronic Structure and Spectrum Properties of 8-Hydroxyquinolinato Manganese Complex [J]. Chem. J. Chinese Universities, 2008, 29(6): 1234. |
| [10] | LI Ming-Xia1,2, ZHOU Xin1, PAN Qing-Jiang2, ZHANG Hong-Xing1*, FU Hong-Gang2, SUN Chia-Chung1. Theoretical Studies on Electronic Structures and Spectroscopic Properties of [Ru(Htcterpy)X3]3-[X=NCS, CN, Cl] [J]. Chem. J. Chinese Universities, 2007, 28(12): 2377. |
| [11] | WU Yin1,2, WANG Jin-Feng2, TENG Yun-Lei2, MENG Xiang-Ying1,2, BAO Yong-Li1,2, BO Hua-Ben2, LI Yu-Xin1. TD-DFT Studies on Electronic Structures and Spectrum Properties for Tectorigenin and Tectoridin [J]. Chem. J. Chinese Universities, 2006, 27(11): 2164. |
| [12] | LU Zhen, FENG Yu-Lin, CHEN Zhao-Bin, XIAO Jun-Ping, YAN Wen-Peng, FAN Mei-Gong . Synthesis and Photochromic Properties of Oxazoly Substituted Fulgide [J]. Chem. J. Chinese Universities, 2005, 26(8): 1451. |
| [13] | LIAO Yi, SU Zhong-Min, CHEN Ya-Guang, KAN Yu-He, DUAN Hong-Xia, QIU Yong-Qing, WANG Rong-Shun . TD-DFT Study on Electronic Spectrum Property for Bis(8-hydroxyquinoline) Beryllium and Its Derivatives [J]. Chem. J. Chinese Universities, 2003, 24(3): 477. |
| [14] | LI Dong-Tao, LI Wen, LI Bao-Qing . In situ Diffuse Reflectance FTIR Study on Water in Lignite [J]. Chem. J. Chinese Universities, 2002, 23(12): 2325. |
| [15] | WANG Hui, ZHAO Jing-Quan, YANG Zi-Xuan, ZHANG Jian-Ping, JIANG Li-Jin. The Structure-Function Relationship of Core Complexes in Phycobilisome(Ⅱ) ──Studies on Chromophore-Chromophore Interactions in Four Allophycocyanin Complexes [J]. Chem. J. Chinese Universities, 1997, 18(9): 1517. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||