

Chem. J. Chinese Universities ›› 2020, Vol. 41 ›› Issue (3): 439.doi: 10.7503/cjcu20190701
• Articles • Previous Articles Next Articles
HE Jinlu,LONG Run,FANG Weihai
Received:2019-12-23
Online:2020-03-10
Published:2020-02-07
Contact:
Run LONG,Weihai FANG
Supported by:CLC Number:
TrendMD:
HE Jinlu, LONG Run, FANG Weihai. A-site Cation Effects on Hot Carrier Relaxation in Perovskites by Nonadiabatic Molecular Dynamics Simulations †[J]. Chem. J. Chinese Universities, 2020, 41(3): 439.
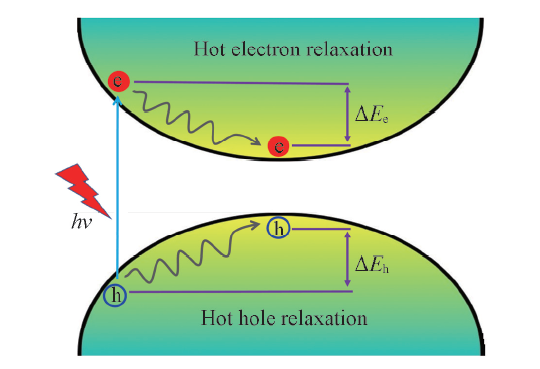
Fig.1 Schematic diagram of hot carrier energy relaxation dynamics in the perovskite system
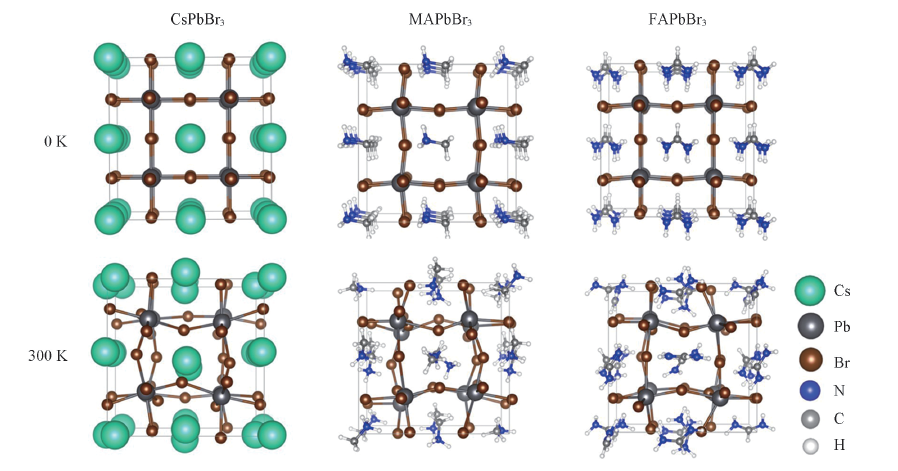
Fig.2 Simulation cell showing the optimized geometry and a representative snapshot of molecular dynamics at 300 K of CsPbBr3, MAPbBr3 and FAPbBr3
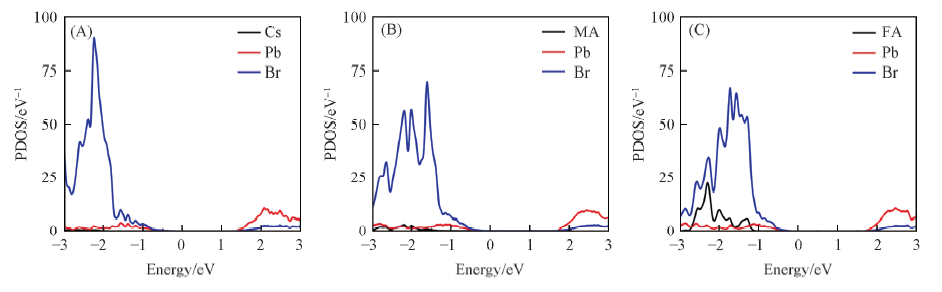
Fig.3 Projected density of states(PDOS) of CsPbBr3(A), MAPbBr3(B) and FAPbBr3(C) systems
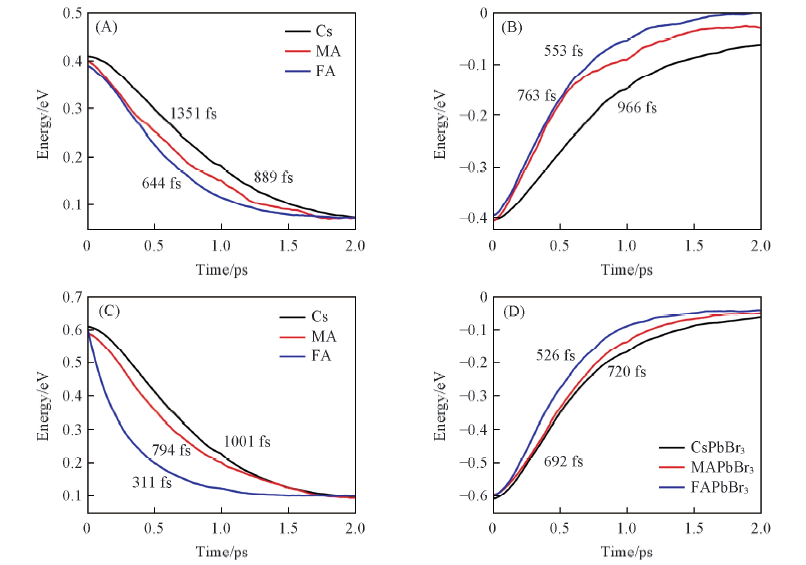
Fig.4 Time evolution of the energy decay dynamics of hot electrons with 0.4 eV(A)and 0.6 eV(C) excess energies, and hot holes with 0.4 eV(B) and 0.6 eV(D) excess energies
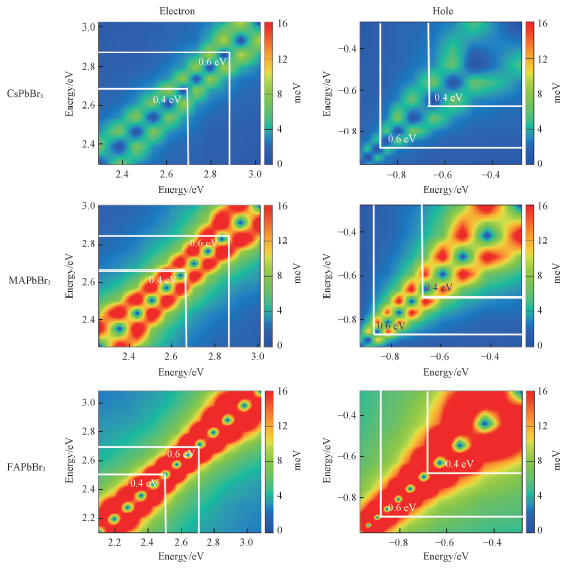
Fig.5 Average magnitude of the nonadiabatic couplings for hot electrons and holes relaxation within conduction band and valence band of the CsPbBr3, MAPbBr3 and FAPbBr3
| [1] | De Wolf S., Holovsky J., Moon S. J., Löper P., Niesen B., Ledinsky M., Haug F. J., Yum J. H., Ballif C ., J. Phys. Chem. Lett., 2014, 5, 1035— 1039 |
| [2] | Shi D., Adinolfi V., Comin R., Yuan M., Alarousu E., Buin A., Chen Y., Hoogland S., Rothenberger A., Katsiev K ., Science, 2015, 347, 519— 522 |
| [3] | Zhu H., Miyata K., Fu Y., Wang J., Joshi P. P., Niesner D., Williams K. W., Jin S., Zhu X. Y ., Science, 2016, 353, 1409— 1413 |
| [4] | Zhu H., Fu Y., Meng F., Wu X., Gong Z., Ding Q., Gustafsson M. V., Trinh M. T., Jin S., Zhu X. Y ., Nature Materials, 2015, 14, 636— 642 |
| [5] | Wang J., Dong J., Lu F., Sun C., Zhang Q., Wang N ., J. Mater. Chem. A, 2019, 7, 23563— 23576 |
| [6] | Azpiroz J. M., Mosconi E., Bisquert J., de Angelis F ., Energy Environ. Sci., 2015, 8, 2118—2127 |
| [7] | Dong X., Fang X., Lü M. H., Lin B. C., Zhang S., Wang Y., Yuan N. Y., Ding J. N ., Chinese Science Bulletin, 2016,1025 |
| ( 董旭, 房香, 吕明航, 林本才, 张帅, 王莹, 袁宁一, 丁建宁 . 科学通报, 2016,1025) | |
| [8] | Chen C., Yang X. C., Liu W., ., CIESC J., 2017, 811— 820 |
| ( 陈超, 杨修春, 刘巍 . 化工学报, 2017, 811— 820) | |
| [9] | Gan Y. S., Chen M. M., Wang Y. L., Wan L., Kong M. Q., Hu H., Wang S. M ., Materials Reports, 2018, 32, 4047—4050, 4078 |
| ( 甘一升, 陈苗苗, 王玉龙, 万丽, 孔梦琴, 胡航, 王世敏 . 材料导报, 2018, 32, 4047—4050, 4078) | |
| [10] | Zhang D. F., Zheng L. L., Ma Y. Z., Wang S. F., Bian Z. Q., Huang C. H., Gong Q. H., Xiao L. X ., Acta Physica Sinica, 2015, 64, 118— 124 |
| ( 张丹霏, 郑灵灵, 马英壮, 王树峰, 卞祖强, 黄春辉, 龚旗煌, 肖立新 . 物理学报, 2015, 64, 118— 124) | |
| [11] | Kojima A., Teshima K., Shirai Y., Miyasaka T ., J. Am. Chem. Soc., 2009, 131, 6050— 6051 |
| [12] | NREL, Efficiency Chart., 2019, |
| [13] | Rühle S ., Sol. Energy, 2016, 130, 139— 147 |
| [14] | Jeon N. J., Noh J. H., Yang W. S., Kim Y. C., Ryu S., Seo J., Seok S. I ., Nature, 2015, 517, 476— 480 |
| [15] | Fu J., Xu Q., Han G., Wu B., Huan C. H. A., Leek M. L., Sum T. C ., Nat. Commun., 2017, 8, 1300 |
| [16] | Chung H., Jung S. I., Kim H. J., Cha W., Sim E., Kim D., Koh W. K., Kim J ., Angew. Chem. Int. Ed. Engl., 2017, 56, 4160— 4164 |
| [17] | Chen J., Messing M. E., Zheng K., Pullerits T ., J. Am. Chem. Soc., 2019, 141, 3532— 3540 |
| [18] | Fang H. H., Adjokatse S., Shao S., Even J., Loi M. A ., Nat. Commun., 2018, 9, 243 |
| [19] | Wang Y., Long R ., J. Phys. Chem. Lett., 2019, 10, 4354— 4361 |
| [20] | Qiao L., Sun X., Long R ., J. Phys. Chem. Lett., 2019, 10, 672— 678 |
| [21] | He J., Fang W. H., Long R., Prezhdo O. V ., ACS Energy Lett., 2018, 3,2070—2076 |
| [22] | Hopper T. R., Gorodetsky A., Frost J. M., Müller C., Lovrincic R., Bakulin A. A ., ACS Energy Lett., 2018, 3, 2199— 2205 |
| [23] | Petersilka M., Gossmann U., Gross E ., Phys. Rev. Lett., 1996, 76, 1212— 1215 |
| [24] | Tully J. C ., J. Chem. Phys., 1990, 93, 1061— 1071 |
| [25] | Craig C. F., Duncan W. R., Prezhdo O. V ., Phys. Rev. Lett., 2005, 95, 163001 |
| [26] | Fischer S. A., Habenicht B. F., Madrid A. B., Duncan W. R., Prezhdo O. V ., J. Chem. Phys., 2011, 134, 024102 |
| [27] | Kohn W., Sham L. J ., Phys. Rev., 1965, 140, A1133— A1138 |
| [28] | Isborn C. M., Li X., Tully J. C ., J. Chem. Phys., 2007, 126, 134307 |
| [29] | Hammes-Schiffer S., Tully J. C ., J. Chem. Phys., 1994, 101, 4657— 4667 |
| [30] | Parandekar P. V., Tully J. C ., J. Chem. Phys., 2005, 122, 094102 |
| [31] | Fabiano E., Keal T., Thiel W ., Chem. Phys., 2008, 349, 334— 347 |
| [32] | Kapral R., Ciccotti G ., J. Chem. Phys., 1999, 110, 8919— 8929 |
| [33] | Herman M. F., Kluk E ., Chem. Phys., 1984, 91, 27— 34 |
| [34] | Tully J ., Faraday Discuss., 1998, 110, 407— 419 |
| [35] | Akimov A. V., Prezhdo O. V ., J. Chem. Theory Comput., 2013, 9, 4959— 4972 |
| [36] | Wang S., Luo Q., Fang W. H., Long R ., J. Phys. Chem. Lett., 2019, 10, 1234— 1241 |
| [37] | Long R., Prezhdo O. V ., Nano Lett., 2014, 14, 3335— 3341 |
| [38] | Li W., Sun Y. Y., Li L., Zhou Z., Tang J., Prezhdo O. V ., J. Am. Chem. Soc., 2018, 140, 15753— 15763 |
| [39] | Long R., Prezhdo O. V ., Nano Lett., 2015, 15, 4274— 4281 |
| [40] | Kresse G., Furthmüller J ., Phys. Rev. B, 1996, 54, 11169— 11186 |
| [41] | Perdew J. P., Burke K., Ernzerhof M ., Phys. Rev. Lett., 1996, 77, 3865— 3868 |
| [42] | Blöchl P. E ., Phys. Rev. B, 1994, 50, 17953— 17979 |
| [43] | Monkhorst H. J., Pack J. D ., Phys. Rev. B, 1976, 13, 5188— 5192 |
| [44] | Grimme S., Antony J., Ehrlich S., Krieg H ., J. Chem. Phys., 2010, 132, 154104 |
| [45] | Rodová M., Brožek J., Knížek K., Nitsch K ., J. Therm. Anal. Calorim., 2003, 71, 667— 673 |
| [46] | Elbaz G. A., Straus D. B., Semonin O. E., Hull T. D., Paley D. W., Kim P., Owen J. S., Kagan C. R., Roy X ., Nano Lett., 2017, 17, 1727— 1732 |
| [47] | Zhumekenov A. A., Saidaminov M. I., Haque M. A., Alarousu E., Sarmah S. P., Murali B., Dursun I., Miao X. H., Abdelhady A. L., Wu T ., ACS Energy Lett., 2016, 1, 32— 37 |
| [48] | Knop O. W., Wasylishen R. E., White M. A., Cameron T. S., Oort M. J. V ., Can. J. Chem., 1990, 68, 412— 422 |
| [49] | Sun S., Isikgor F. H., Deng Z., Wei F., Kieslich G., Bristowe P. D., Ouyang J., Cheetham A. K ., ChemSusChem, 2017, 10, 3740— 3745 |
| [50] | Jankowska J., Prezhdo O. V ., J. Phys. Chem. Lett., 2017, 8, 812— 818 |
| [51] | Motta C., El-Mellouhi F., Sanvito S ., Phys. Rev. B, 2016, 93, 235412 |
| [52] | El-Mellouhi F., Marzouk A., Bentria E. T., Rashkeev S. N., Kais S., Alharbi F. H ., ChemSusChem, 2016, 9, 2648— 2655 |
| [53] | Ye Y., Run X., Xu H. T., Feng H., Fei X., Wang L. J ., Chin. Phys. B, 2015, 24, 116302— 116303 |
| [54] | Tavernelli I., Tapavicza E., Rothlisberger U ., J. Chem. Phys., 2009, 130, 124107 |
| [1] | JIA Yanggang, SHAO Xia, CHENG Jie, WANG Pengpeng, MAO Aiqin. Preparation and Lithium Storage Performance of Pseudocapacitance-controlled Perovskite High-entropy Oxide La(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)O3 Anode Materials [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220157. |
| [2] | YUAN Meng, ZHAO Yingjie, WU Yuchen, JIANG Lei. Assembly of Perovskite Arrays and Multifunctional Detector Applications [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220448. |
| [3] | GAO Zhongnan, GUO Lihong, ZHAO Dongyue, LI Xingang. Effect of A Site-deficiency on the Structure and Catalytic Oxidation Activity of the La-Sr-Co-O Perovskite [J]. Chem. J. Chinese Universities, 2021, 42(9): 2869. |
| [4] | YUE Shengli, WU Guangbao, LI Xing, LI Kang, HUANG Gaosheng, TANG Yi, ZHOU Huiqiong. Research Progress of Quasi-two-dimensional Perovskite Solar Cells [J]. Chem. J. Chinese Universities, 2021, 42(6): 1648. |
| [5] | LI Yishan, GUO Liang, PENG Sifan, ZHANG Qingmao, ZHANG Yuhao, XU Shiqi. Cobalt Substitutions in Lanthanum Manganate Photocatalyst: First-principles and Visible-light Photocatalytic Ability Investigation [J]. Chem. J. Chinese Universities, 2021, 42(6): 1881. |
| [6] | WANG Kunhua, YAO Jisong, YANG Junnan, SONG Yonghui, LIU Yuying, YAO Hongbin. Synthesis and Device Optimization of Highly Efficient Metal Halide Perovskite Light-emitting Diodes [J]. Chem. J. Chinese Universities, 2021, 42(5): 1464. |
| [7] | DONG Luming, SU Yanyue, WANG Chunzheng, QIAO Yafei, CHEN Yajun, MA Haiyun. Synthesis of Micro- to Nano-scale Perovskite Calcium Hydroxytinate and Its Performance as a Flame Retardant in Epoxy Resin [J]. Chem. J. Chinese Universities, 2021, 42(3): 937. |
| [8] | LIU Yao, DENG Zhengtao. Fast Synthesis of Highly Luminescent Two-dimensional Tin-halide Perovskites by Anti-solvent Method [J]. Chem. J. Chinese Universities, 2021, 42(12): 3774. |
| [9] | WANG Tingting, LEI Yuhan, LIN Yujuan, HUANG Jialing, LIU Cuie, ZHENG Fengying, LI Shunxing. Preparation of Liposome-terminated CsPbX3(X=Cl,Br,I) Nanocrystals and Applications in Light-emitting Diode Devices [J]. Chem. J. Chinese Universities, 2020, 41(8): 1896. |
| [10] | CHENG Rongmin,XU Hong,SHAN Ruiping,ZHAN Conghong. Influential Factors of La-doped Calcium Titanate for Photocatalytic H2 Evolution Under Visible Light † [J]. Chem. J. Chinese Universities, 2020, 41(6): 1345. |
| [11] |
HAN Hongjing,GE Qin,CHEN Yanguang,WANG Haiying,ZHAO Hongzhi,WANG Yizhen,ZHANG Yanan,DENG Jitong,SONG Hua,ZHANG Mei.
Production of Phenolic Compounds from Bagasse Lignin via Catalytic Pyrolysis of Ca1-xPrxFe |
| [12] | JIANG Yefang, DONG Ru, CAI Xuediao, FENG Jiangshan, LIU Zhike, LIU Shengzhong. Effect of Amphiphilic Quaternary Ammonium Salt Additive on Performance and Stability of Perovskite Solar Cells [J]. Chem. J. Chinese Universities, 2019, 40(8): 1697. |
| [13] | JI Tianhao, TIAN Yanqing. Optical Properties and Applications of Halide-perovskite Nanocrystals† [J]. Chem. J. Chinese Universities, 2018, 39(6): 1113. |
| [14] | LIU Guozhen, YE Jiajiu, HOU Zhaosheng, CHEN Shuanghong, HU Linhua, PAN Xu, DAI Songyuan. Influence of Crack-defect on Perovskite Solar Cells Performance† [J]. Chem. J. Chinese Universities, 2018, 39(3): 545. |
| [15] | LIU Yang, FU Xianwei, ZHAO Tianyu, LIAN Gang, DONG Ning, SONG Side, WANG Qilong, CUI Deliang. Investigation on Stability of Perovskite Semiconductor CH3NH3PbI3 by In-situ FTIR Spectroscopy† [J]. Chem. J. Chinese Universities, 2016, 37(9): 1605. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||