高等学校化学学报
2026, 47 (
):
20250266-.
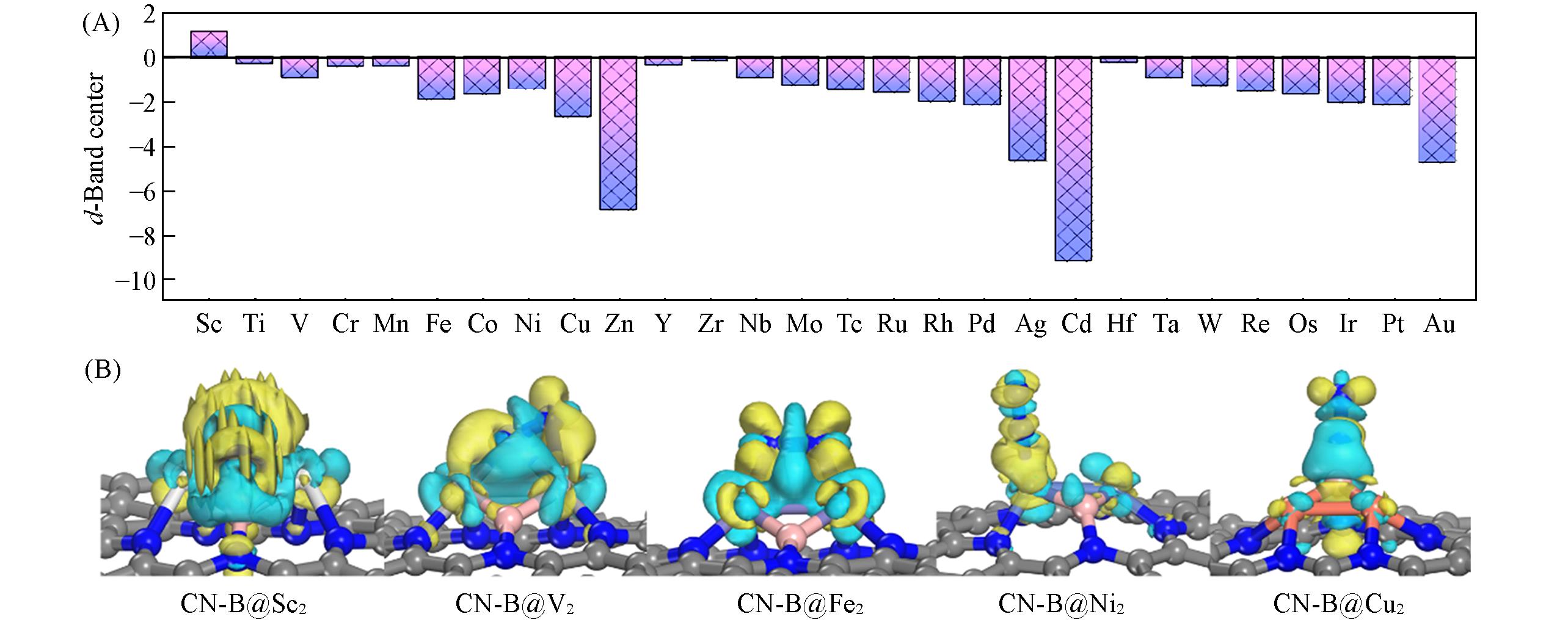
通过高通量密度泛函计算筛选出一系列具有氮还原反应(NRR)活性的B和双金属原子组成的CN-B@M2催化剂. CN-B@Fe2, CN-B@Tc2, CN-B@Os2和CN-B@Re2被认为是具有良好选择性和NRR活性的催化剂, 其极限电位(UL)分别为-0.24, -0.34, -0.31和-0.38 V. 计算结果表明, N2在B@M2上的吸附呈周期性演变, 吸附构型和能量受d带中心调节. UL随转移电荷呈火山型分布. 具有中等电子给体能力(中等电荷转移)的B@M2催化剂表现出优异的NRR活性. 通过量化催化剂的原子电子特性和拓扑结构, 构建了用于描述给电子能力的描述 符Φ. 结果表明, 给电子能力与氮还原反应的极限电位呈火山关系. 使用描述符Φ和催化剂的内在特性作为特征预测了吸附能和极限电位, 由于R2值为0.99, 梯度提升回归(GBR)被认为是构建机器学习预测模型的最恰当方法.





{kind=link}