高等学校化学学报
2023, 44 (
):
20220721-.
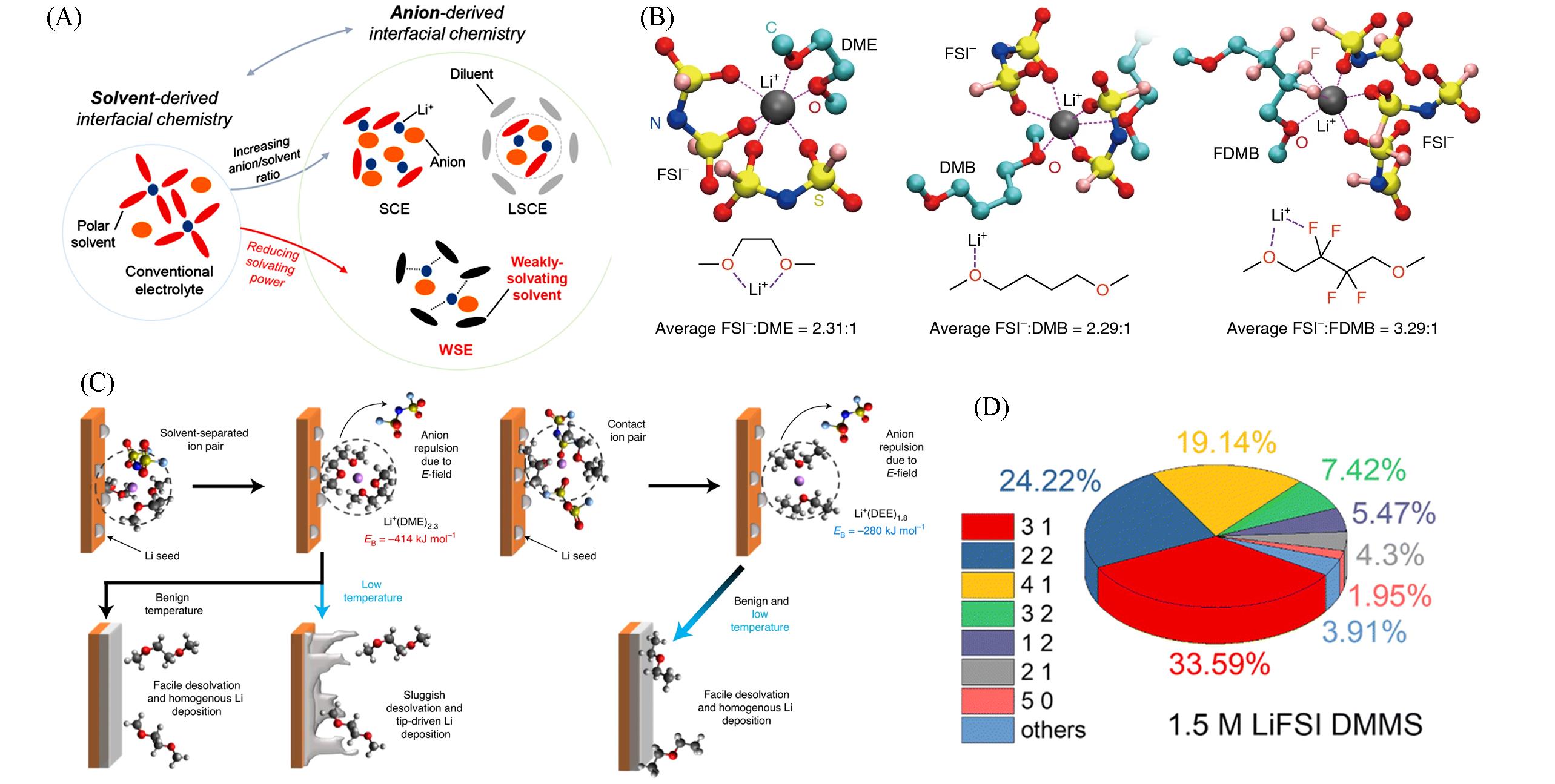
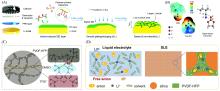
金属锂负极具有极高的理论比容量和极低的氧化还原电位, 被认为是二次电池体系中负极材料的最终选择. 但在实际应用过程中, 不稳定的电极/电解液界面会造成大量的锂枝晶生长, 导致容量损失乃至热失控等安全问题. 调控锂离子溶剂化结构, 可促进有益的固态电解质界面膜(SEI)成膜组分在电极表面优先分解, 进而稳定电极界面并可诱导锂离子均匀沉积, 是提升液态和准固态金属锂电池电化学性能的重要手段. 本文综合评述了近年来从液态到准固态电解质中锂离子溶剂化结构调控的策略和设计原则, 探讨了溶剂化结构改变对电极/电解质界面的影响, 并对准固态电解质的研究前景进行了展望.



{kind=link}