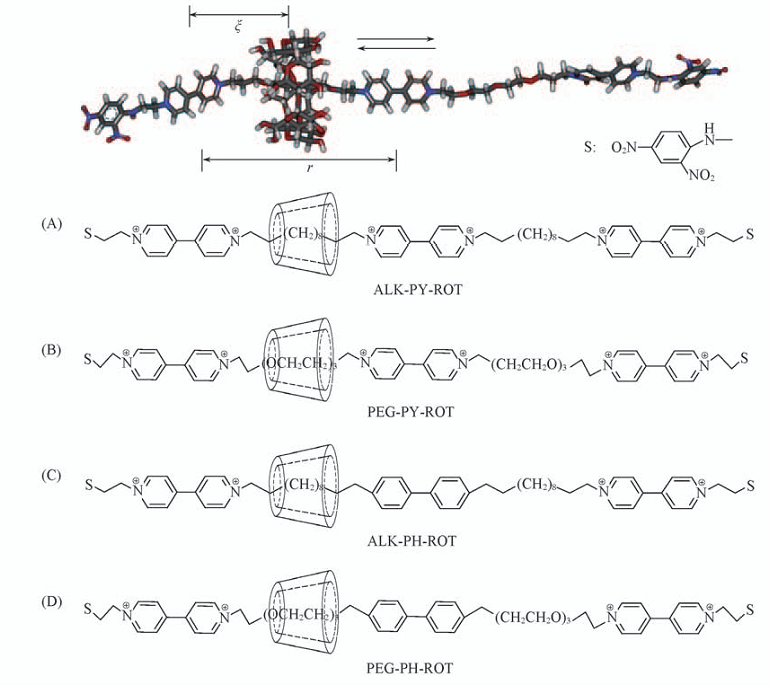
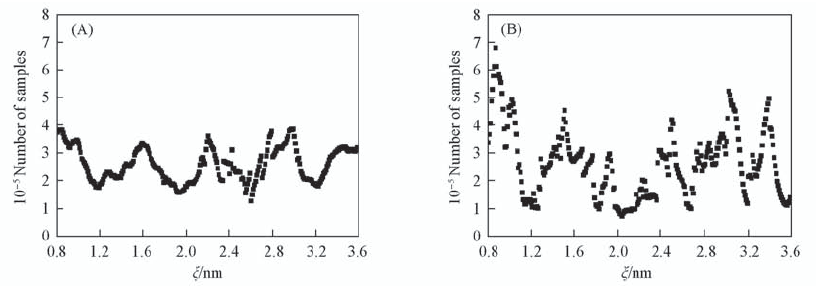
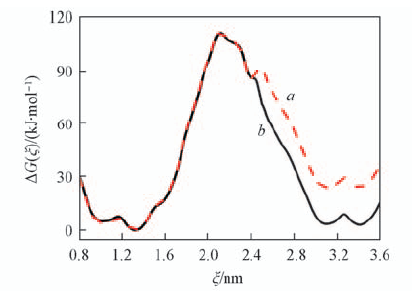
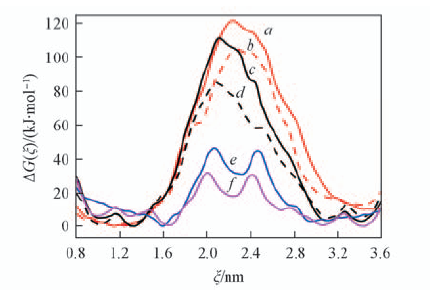
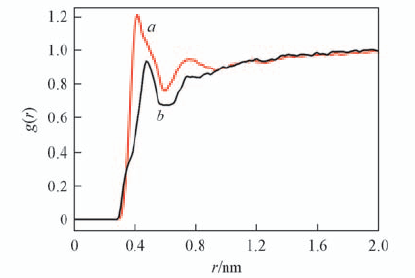
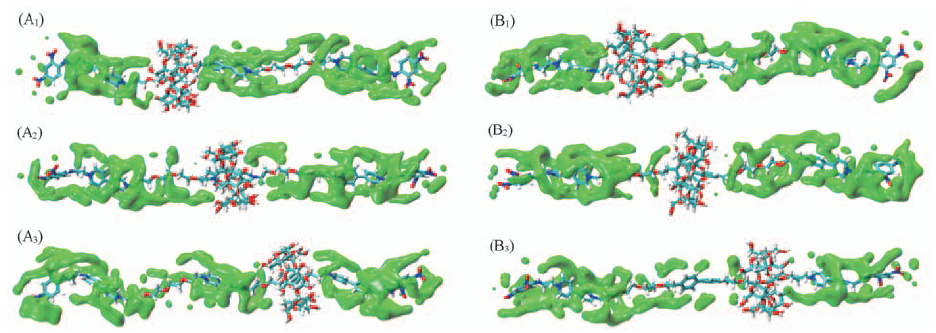
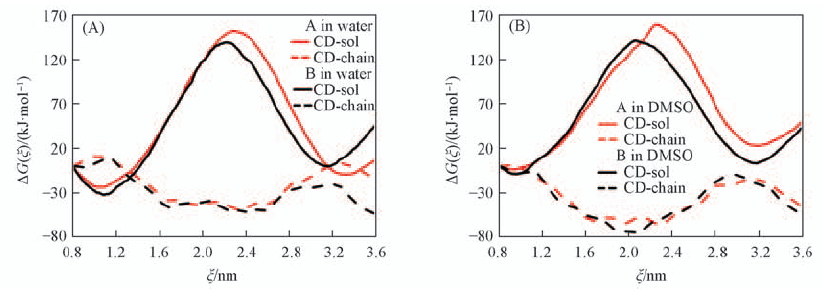
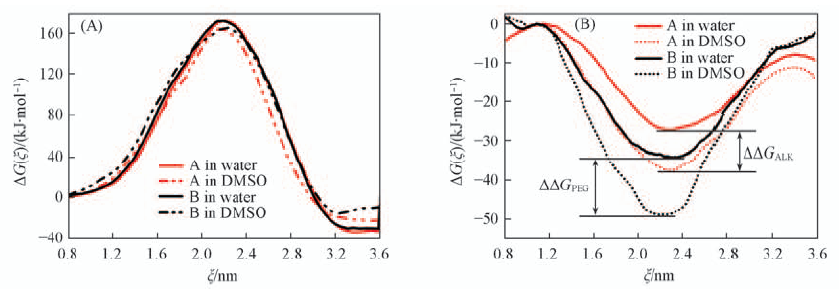
| [1] |
Zhai C. X., Huang F. H., Sci. China, Ser. B: Chem., 2009, 39(4), 315—328
|
|
(翟春熙, 黄飞鹤. 中国科学B辑: 化学, 2009, 39(4), 315—328)
|
| [2] |
Ma X., Tian H., Acc. Chem. Res., 2014, 47(7), 1971—1981
|
| [3] |
Sun S., Shi J. B., Dong Y. P., Hu X. Y., Wang L. Y., Prog. Chem., 2014, 26(8), 1409—1426
|
|
(孙书, 石建兵, 董宇平, 胡晓玉, 王乐勇. 化学进展, 2014, 26(8), 1409—1426)
|
| [4] |
Yang N. W., Chen T., Fu J. J., Chem. J. Chinese Universities, 2014, 35(5), 971—975
|
|
(杨年旺, 陈涛, 傅佳骏. 高等学校化学学报, 2014, 35(5), 971—975)
|
| [5] |
Liu P., Shao X. G., Cai W. S., Prog. Chem., 2013, 25(5), 692—697
|
|
(刘鹏, 邵学广, 蔡文生. 化学进展, 2013, 25(5), 692—697)
|
| [6] |
Gao P., Wang P. J., Geng X., Ye L., Zhang A. Y., Feng Z. G., Acta Chim. Sinica, 2013, 71(3), 347—350
|
|
(高鹏, 王培境, 耿雪, 叶霖, 张爱英, 冯增国. 化学学报, 2013, 71(3), 347—350)
|
| [7] |
Han B., Liao X. L., Yang B., Prog. Chem., 2014, 26(6), 1039—1049
|
|
(韩彬, 廖霞俐, 杨波. 化学进展, 2014, 26(6), 1039—1049)
|
| [8] |
Ma X., Tian H., Chem. Soc. Rev., 2010, 39, 70—80
|
| [9] |
Murakami H., Kawabuchi A., Matsumoto R., Ido T., Nakashima N., J. Am. Chem. Soc., 2005, 127(45), 15891—15899
|
| [10] |
Wenz G., Han B. H., Müller A., Chem. Rev., 2006, 106(3), 782—817
|
| [11] |
Kawaguchi Y., Harada A., Org. Lett., 2000, 2(10), 1353—1356
|
| [12] |
Liu P., Chipot C., Shao X. G., Cai W. S., J. Phys. Chem. C, 2012, 116(7), 4471—4476
|
| [13] |
Hénin J., Chipot C., J. Chem. Phys., 2004, 121(7), 2904—2914
|
| [14] |
Chipot C., Hénin J., J. Chem. Phys., 2005, 123(24), 244906
|
| [15] |
Rodriguez-Gomez D., Darve E., Pohorille A., J. Chem. Phys., 2004, 120(8), 3563—3578
|
| [16] |
Comer J., Roux B., Chipot C., Mol. Simulat., 2014, 40(1—3), 218—228
|
| [17] |
Comer J., Gumbart J. C., Hénin J., Lelièvre T., Pohorille A., Chipot C., J. Phys. Chem. B, 2015, 119(3), 1129—1151
|
| [18] |
Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., J. Comput. Chem., 2005, 26(16), 1781—1802
|
| [19] |
Kuttel M., Brady J. W., Naidoo K. J., J. Comput. Chem., 2002, 23(13), 1236—1243
|
| [20] |
Best R. B., Zhu X., Shim J. Y., Lopes P. E. M., Mittal J., Feig M., Mackerell A. D., J. Chem. Theory Comput., 2012, 8(9), 3257—3273
|
| [21] |
Liu P., Cai W. S., Chipot C., Shao X. G., J. Phys. Chem. Lett., 2010, 1(12), 1776—1780
|
| [22] |
Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys., 1983, 79(2), 926—935
|
| [23] |
Strader M. L., Feller S. E., J. Phys. Chem. A, 2002, 106(6), 1074—1080
|
| [24] |
Feller S. E., Zhang Y. H., Pastor R. W., Brooks B. R., J. Chem. Phys., 1995, 103(11), 4613—4621
|
| [25] |
Ryckaert J. P., Ciccotti G., Berendsen H. J. C., J. Chem. Phys., 1977, 23(3), 327—341
|
| [26] |
Andersen H. C., J. Chem. Phys., 1983, 52(1), 24—34
|
| [27] |
Miyamoto S., Kollman P. A., J. Comput. Chem., 1992, 13(8), 952—962
|
| [28] |
Darden T., York D., Pedersen L., J. Chem. Phys., 1993, 98(12), 10089—10092
|
| [29] |
Tuckerman M., Berne B. J., Martyna G. J., J. Chem. Phys., 1992, 97(3), 1990—2001
|
| [30] |
Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 1996, 14(1), 33—38
|
| [31] |
Hénin J., Fiorin G., Chipot C., Klein M. L., J. Chem. Theory Comput., 2010, 6(1), 35—47
|
| [32] |
Eyring H., J. Chem. Phys., 1935, 3, 107—115
|
| [33] |
Collet O., Chipot C., J. Am. Chem. Soc., 2003, 125(21), 6573—6580
|
 )
)