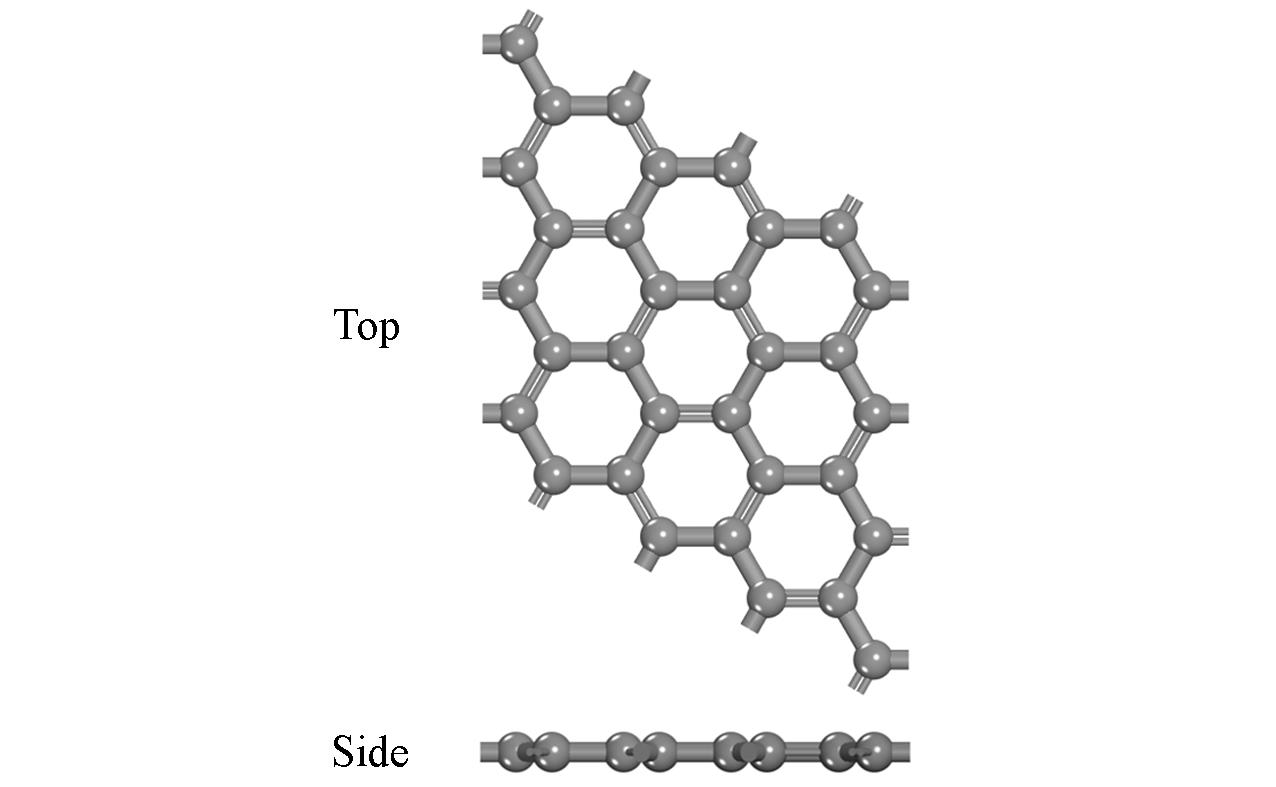
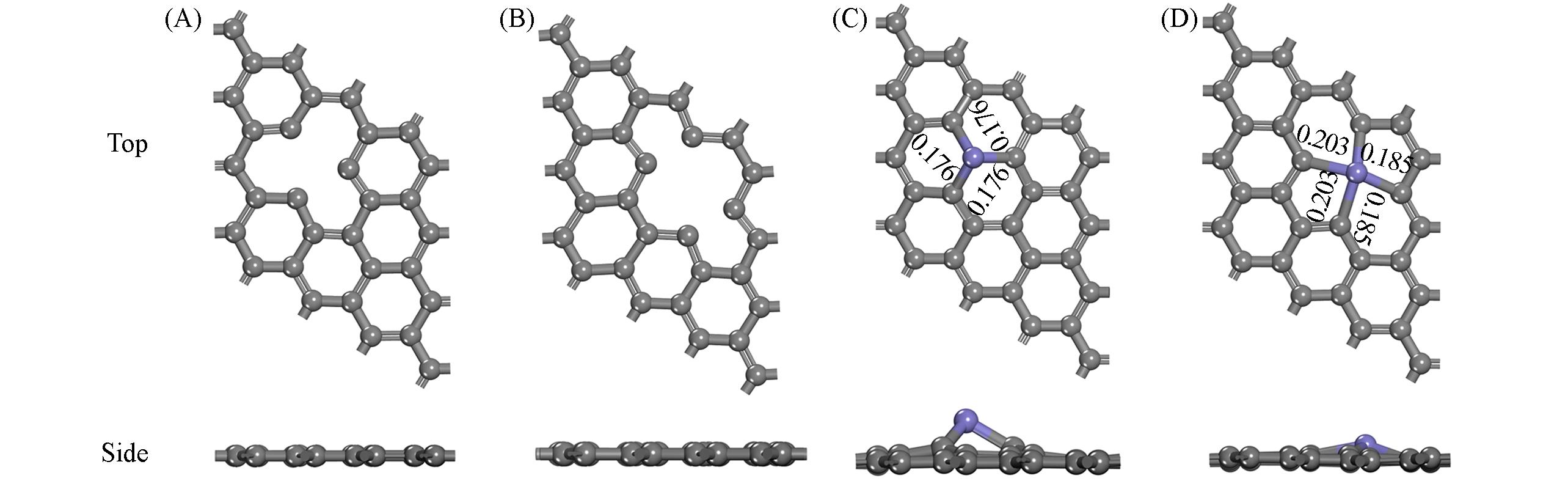
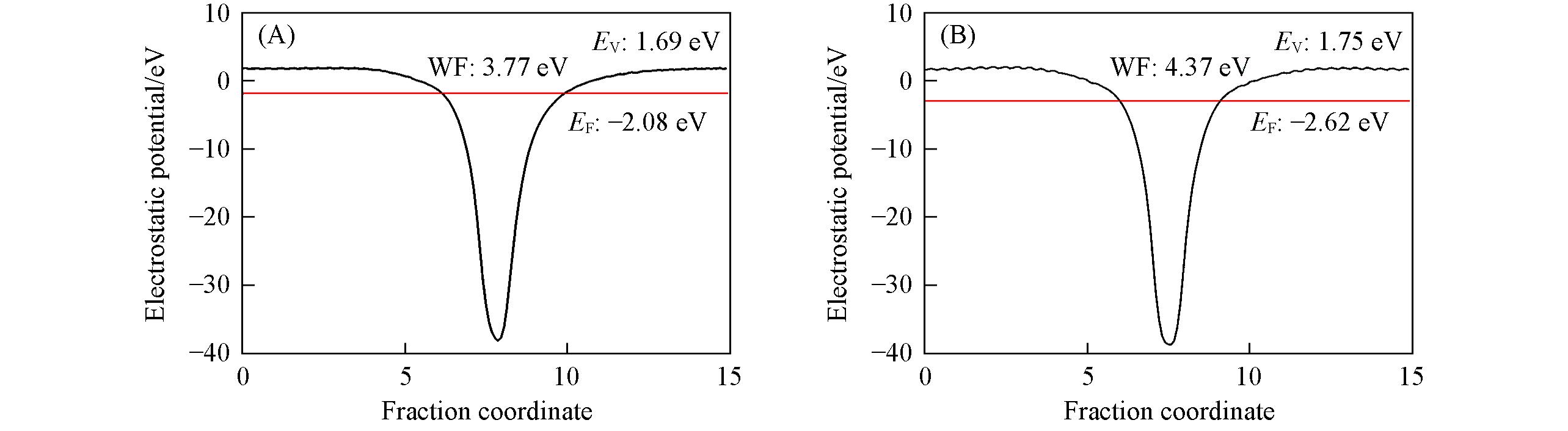
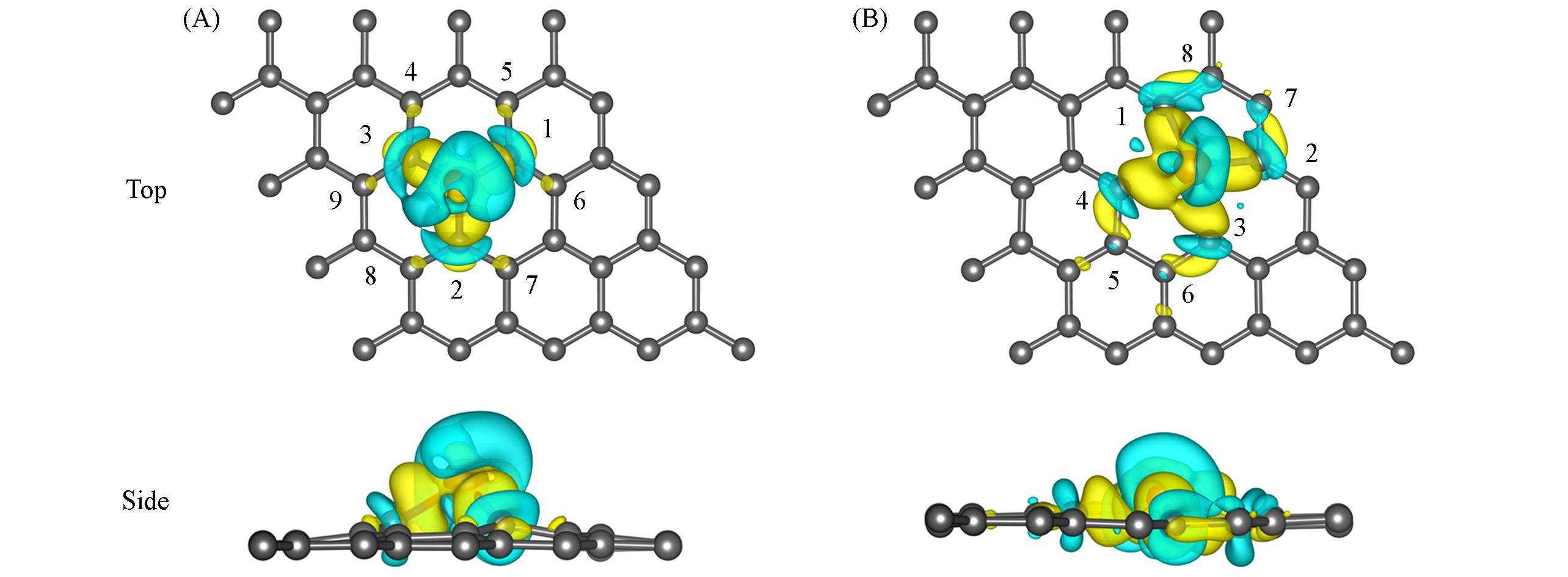
| [1] |
Fischer F., Tropsch H., Brennst. Chem., 1923, 4, 276—285
|
| [2] |
Fischer F., Tropsch H., Brennst. Chem., 1926, 7, 97—104
|
| [3] |
Brady R. C., Pettit R., J. Am. Chem. Soc., 1981, 103, 1287—1289
|
| [4] |
Venter J., Kaminsky M., Geoffroy G. L., Albert V. M., J. Catal., 1987, 103, 450—465
|
| [5] |
Smit E. D., Weckhuysen B. M., Chem. Soc. Rev., 2008, 37, 2758—2781
|
| [6] |
Ramoshibidu P. M., Nico F., Michael C., Eric Van S., J. Catal., 2012, 289, 140—150
|
| [7] |
Smit E. D., Cinquini F., Beale A. M., Safonova O. V., Van Beek W., Sautet P., Weckhuysen B. M., J. Am. Chem. Soc., 2010, 132, 14928—14941
|
| [8] |
Niemantsverdriet J. W., Van Der Kraan A. M., Van Dijk W. L., Van Der Ban H. S., J. Phys. Chem., 1980, 84, 3363—3370
|
| [9] |
Commereuc D., Chauvin Y., Hugue F., Basset J. M., Olivier D., J. Chem. Soc. Chem. Commun., 1980, 4, 2154—2155
|
| [10] |
Zhang C., Yang Y., Teng B., Li T., Zheng H., Xiang H., Li Y., J. Catal., 2006, 237, 405—415
|
| [11] |
Sun B., Xu K., Nguyen L., Qiao M., Tao F., ChemCatChem, 2012, 4(10), 1498—1511
|
| [12] |
Torres G. H. M., Bitter J. H., Davidian T., Ruitenbeek M., Dugulan A. I., Krijn P., De J. K. P., J. Am. Chem. Soc., 2012, 134, 16207—16215
|
| [13] |
Schutle H. J., Graf B., Xia W., Muhler M., ChemCatChem, 2012, 4, 350—355
|
| [14] |
Abbaslou R. M. M., Soltan J., Dalai A. K., Appl. Catal. A, 2010, 379, 129—134
|
| [15] |
Cheng Y., Lin J., Xu K., Wang H., Yao X., Pei Y., Yan S., Qiao M., Zong B., ACS Catal., 2016, 6, 389—399
|
| [16] |
Wei Y., Luo D., Zhang C., Liu J., He Y., Wen X., Yang Y., Li Y., Catal. Sci. Technol., 2018, 8, 2883—2893
|
| [17] |
Zhang Z., Wei Y., Yu J, Ren M., Zhao C., Chen F., Zhang C., Jiang Y., Guo L., Sun S., ACS Sustainable Chem. Eng., 2024, 12, 17158—17166
|
| [18] |
Zhao H., Zhu Q., Gao Y., Zhai P., Ma D., Appl. Catal. A, 2013, 456, 233—239
|
| [19] |
Wei Y., Yan L., Ma C., Zhang C., Sun S., Wen X., Yang Y., Li Y., ACS Appl. Nano Mater., 2020, 3, 7182—7191
|
| [20] |
Zhang X. Q., Li Z., Sun W., Zhang Y. H., Li J. L.,Wang L., Proc. Natl. Acad. Sci. USA, 2024, 121(50), e2407624121
|
| [21] |
Wang J., Song Y. Y., Chen C., Zhao X., Fan W. L., ACS Catal., 2023, 13, 15794—15810
|
| [22] |
Lin Y. F., Wang Y., Weng Z. L., Zhou Y., Liu S. Q., Ou X. W., Xu X., Cai Y. P., Jiang J., Han B., Yang Z. F., Nat. Commun., 2024, 15, 10032
|
| [23] |
Tang C., Jiao Y., Shi B. Y., Liu N. J., Xie Z. H., Chen X., Zhang Q., Qiao S., Angew. Chem. Int. Ed., 2020, 59, 9171—9176
|
| [24] |
Kresse G., Furthmüller J., Comp. Mater. Sci., 1996, 6, 15—50
|
| [25] |
Kresse G., Furthmüller J., Phys. Rev. B, 1996, 54, 11169—11186
|
| [26] |
Blochl P. E., Phys. Rev. B, 1994, 50, 17953—17979
|
| [27] |
Kresse G., Phys. Rev. B, 1999, 59, 1758—1775
|
| [28] |
Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett., 1996, 77, 3865—3868
|
| [29] |
Henkelman G., Jonsson H., J. Chem. Phys., 2000, 113, 9978—9985
|
| [30] |
Luo D., Ren P. J., Liu X. C., Gao R., Zhou Y. W., Guo W. P., Yang Y., Li Y. W., Wen X. D., J. Phys. Chem. C, 2018, 122, 24037—24045
|
| [31] |
Majidi R., Theor. Chem. Acc., 2017, 136, 109
|
| [32] |
Wang H. T., Wang Q. X., Cheng Y. C., Li K., Yao Y. B., Zhang Q., Dong C. Z., Wang P., Schwingenschlögl U., Yang W., Zhang X. X., Nano. Lett., 2012, 12, 141—144
|
| [33] |
Chen J. H., Li L., Cullen W. G., Williams E. D., Fuhrer M. S., Nat. Phys., 2011, 7, 535—538
|
| [34] |
Shankara S. K., Ranveer S., Hyungtak S., Appl. Catal. B: Environ., 2021, 295, 120269
|
| [35] |
Meng Q. Q., Lv C., Sun J. X., Hong W. Z., Xing W. N., Qiang L. S., Chen G., Jin X. L., Appl. Catal. B: Environ., 2019, 256, 117781
|
| [36] |
Yu Y. X., J. Mater. Chem. A, 2014, 2, 8910—8917
|
| [37] |
Ma Z. F., Liu X., Wang X. S., Luo Z. G. Li W. R., Nie Y. H., Pei L., Mao Q. N., Wen X., Zhong J. S., Chem. Eng. J., 2023, 468, 143569
|
| [38] |
Zhang H. M., Zhang Z. Q., Liu Y. M., Fang X. Z., Xu J. W., Wang X., Xu X. L., Phys. Chem. Lett., 2021, 12, 9188—9196
|
| [39] |
Yu Y. X., J. Colloid Interf. Sci., 2025, 695, 137799
|
| [40] |
Huo C. F., Li Y. W., Wang J. G., Jiao H. J., J. Phys. Chem. C, 2008, 112, 14108—14116
|
| [41] |
Zhang R. G., Liu F., Wang Q., Wang B. J., Li D. B., Appl. Catal. A: General., 2016, 525, 76—84
|
 ), WANG Jianxin1, LU Kuan2, WANG Min1, CHANG Tong1(
), WANG Jianxin1, LU Kuan2, WANG Min1, CHANG Tong1(