

Chem. J. Chinese Universities ›› 2023, Vol. 44 ›› Issue (12): 20230372.doi: 10.7503/cjcu20230372
• Physical Chemistry • Previous Articles Next Articles
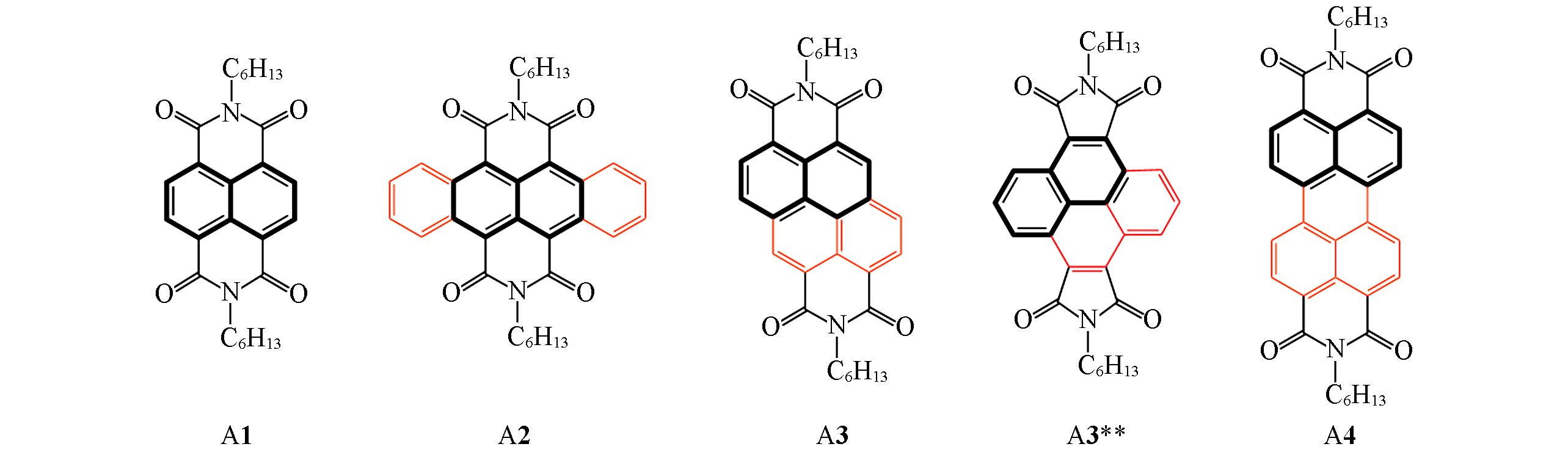
PAN Jiazheng1, SUN Xiaoqi1, REN Aimin2, GUO Jingfu1( )
)
Received:2023-08-17
Online:2023-12-10
Published:2023-10-08
Contact:
GUO Jingfu
E-mail:guojf217@nenu.edu.cn
Supported by:CLC Number:
TrendMD:
PAN Jiazheng, SUN Xiaoqi, REN Aimin, GUO Jingfu. Theoretical Study of the Charge Transport Properties of Naphthalene Tetracarboxylic Diimide Organic Semiconductors Based on Different π-Core Extensions[J]. Chem. J. Chinese Universities, 2023, 44(12): 20230372.
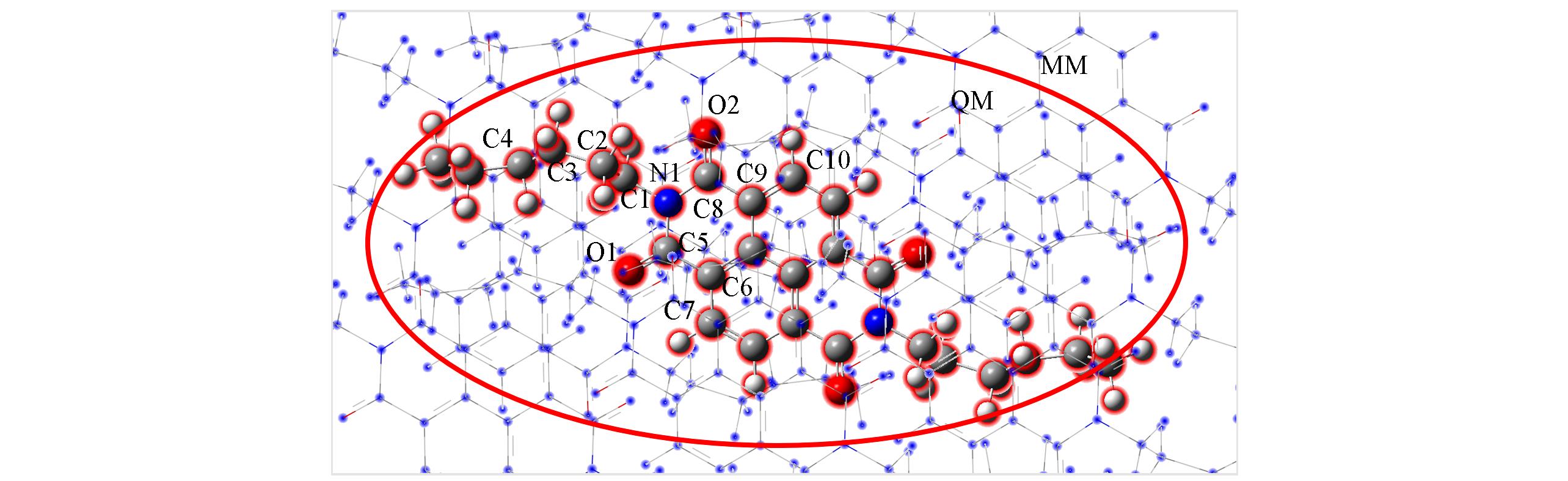
| Molecule | Gas(QM⁃Model) | Solid(QM/MM⁃Model) | X⁃ray | |||
|---|---|---|---|---|---|---|
| α1/(°) | α2/(°) | α1/(°) | α2/(°) | α1/(°) | α2/(°) | |
| A1 | -0.79 | 0.92 | 2.14 | 2.31 | 2.14 | 2.32 |
| A2 | 13.52 | 16.62 | 14.70 | 17.25 | 13.58 | 19.29 |
| A3 | 0.79 | -0.82 | 2.44 | 2.89 | 2.44 | 2.90 |
| A3** | 0.77 | -0.83 | 0.14 | 0.21 | 0.13 | 0.21 |
| A4 | 0.82 | -0.82 | 1.62 | -1.80 | 1.63 | -1.81 |
Table 1 Dihedral angles( α ) between the acyl group and the central benzene ring of the series of molecules*
| Molecule | Gas(QM⁃Model) | Solid(QM/MM⁃Model) | X⁃ray | |||
|---|---|---|---|---|---|---|
| α1/(°) | α2/(°) | α1/(°) | α2/(°) | α1/(°) | α2/(°) | |
| A1 | -0.79 | 0.92 | 2.14 | 2.31 | 2.14 | 2.32 |
| A2 | 13.52 | 16.62 | 14.70 | 17.25 | 13.58 | 19.29 |
| A3 | 0.79 | -0.82 | 2.44 | 2.89 | 2.44 | 2.90 |
| A3** | 0.77 | -0.83 | 0.14 | 0.21 | 0.13 | 0.21 |
| A4 | 0.82 | -0.82 | 1.62 | -1.80 | 1.63 | -1.81 |
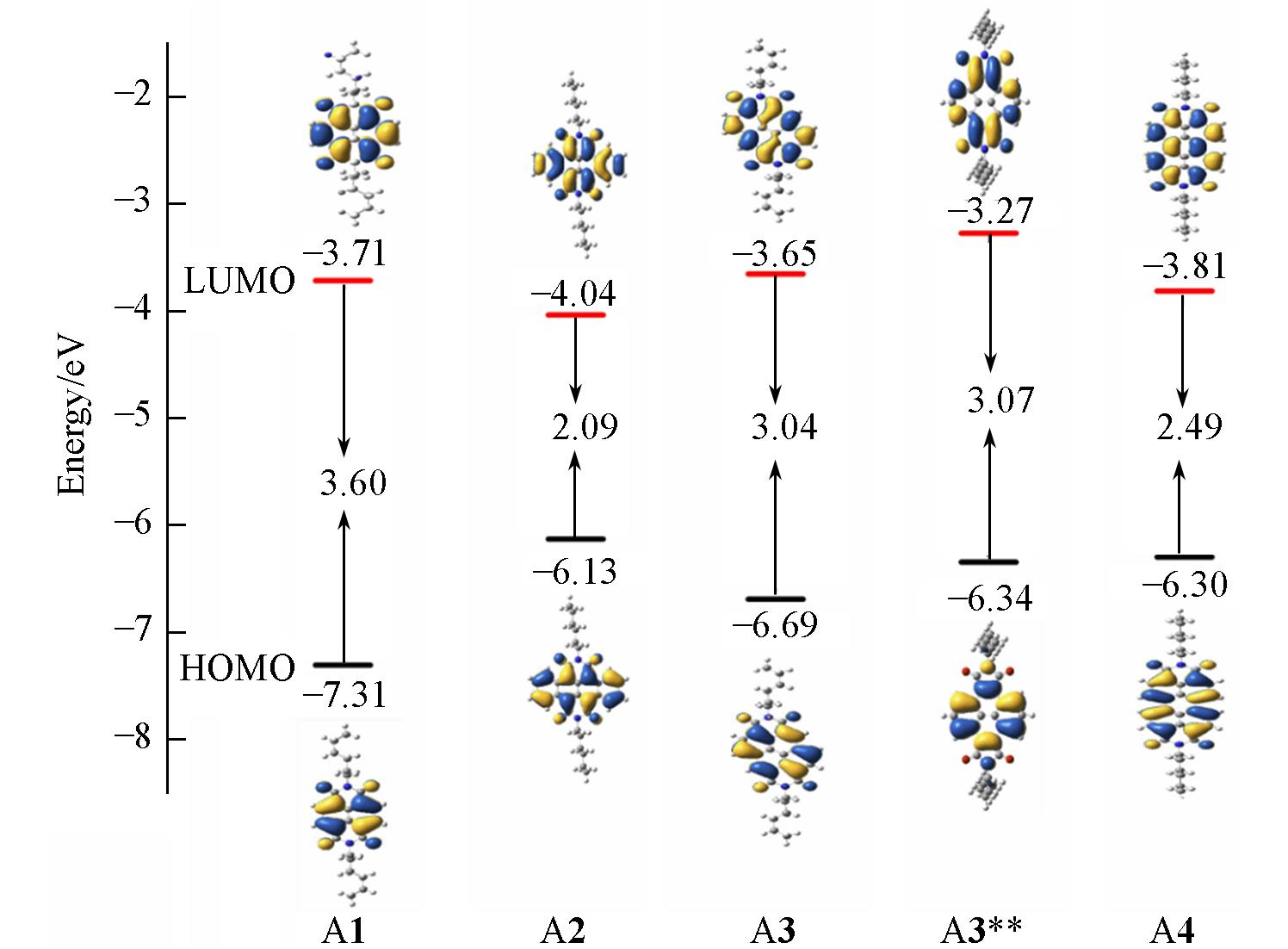
| Molecule | EHOMO/eV | ELUMO/eV | Eg/eV | EHOMO(exp)/eV | ELUMO(exp)/eV | EA(a)/eV |
|---|---|---|---|---|---|---|
| A1 | -7.31 | -3.71 | 3.60 | — | -3.89 | 2.22 |
| A2 | -6.13 | -4.04 | 2.09 | — | -4.13 | 2.54 |
| A3 | -6.69 | -3.65 | 3.04 | -6.43 | -3.69 | 2.11 |
| A3** | -6.34 | -3.27 | 3.07 | — | — | 1.77 |
| A4 | -6.30 | -3.81 | 2.49 | — | — | 2.33 |
| Naphthalene | — | -1.34 | — | — | — | — |
| Tetracene | — | -2.40 | — | — | — | — |
| Pyrene | — | -1.76 | — | — | — | — |
| Perylene | — | -2.19 | — | — | — | — |
Table 2 Calculated electronic structures for the NDI derivatives and reference molecule at the level of B3LYP/6-311++G(d,p)//B3LYP/6-31G(d,p)
| Molecule | EHOMO/eV | ELUMO/eV | Eg/eV | EHOMO(exp)/eV | ELUMO(exp)/eV | EA(a)/eV |
|---|---|---|---|---|---|---|
| A1 | -7.31 | -3.71 | 3.60 | — | -3.89 | 2.22 |
| A2 | -6.13 | -4.04 | 2.09 | — | -4.13 | 2.54 |
| A3 | -6.69 | -3.65 | 3.04 | -6.43 | -3.69 | 2.11 |
| A3** | -6.34 | -3.27 | 3.07 | — | — | 1.77 |
| A4 | -6.30 | -3.81 | 2.49 | — | — | 2.33 |
| Naphthalene | — | -1.34 | — | — | — | — |
| Tetracene | — | -2.40 | — | — | — | — |
| Pyrene | — | -1.76 | — | — | — | — |
| Perylene | — | -2.19 | — | — | — | — |
| Molecule | λAP/eV | λNM/eV | λlow/eV | λhigh/eV |
|---|---|---|---|---|
| A1 | 346.4 | 353.5 | 78.5 | 267.9 |
| A2 | 274.6 | 274.8 | 90.8 | 183.8 |
| A3 | 289.2 | 289.9 | 61.6 | 227.6 |
| A3** | 248.2 | 248.6 | 66.1 | 182.1 |
| A4 | 268.9 | 269.1 | 50.6 | 218.3 |
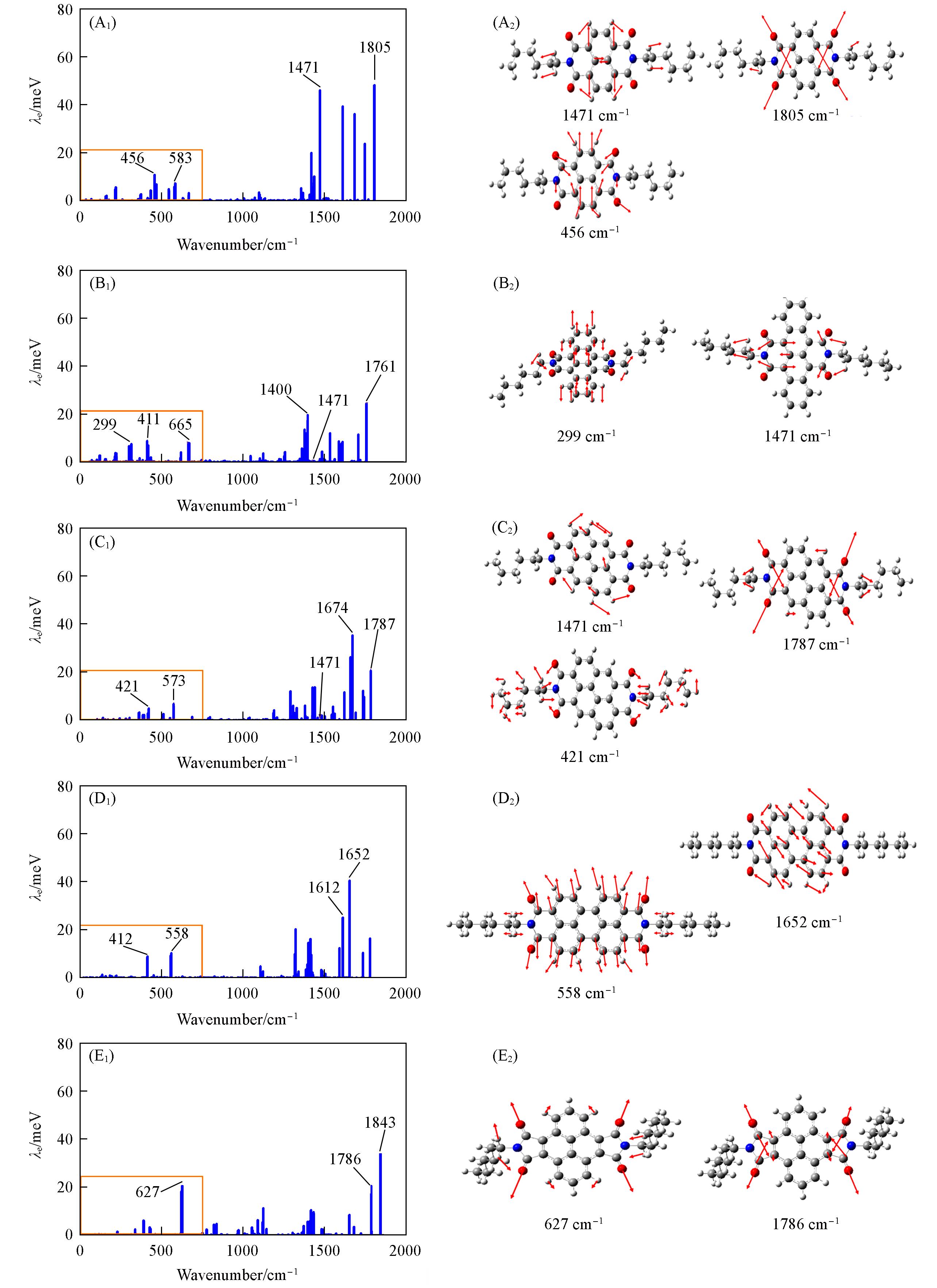
Table 3 Calculated total intramolecular electronic reorganization energies( λAP/ λNM), and distribution in the high and low frequency regions( λlow/ λhigh) with QM/MM model for studied NDI derivatives
| Molecule | λAP/eV | λNM/eV | λlow/eV | λhigh/eV |
|---|---|---|---|---|
| A1 | 346.4 | 353.5 | 78.5 | 267.9 |
| A2 | 274.6 | 274.8 | 90.8 | 183.8 |
| A3 | 289.2 | 289.9 | 61.6 | 227.6 |
| A3** | 248.2 | 248.6 | 66.1 | 182.1 |
| A4 | 268.9 | 269.1 | 50.6 | 218.3 |
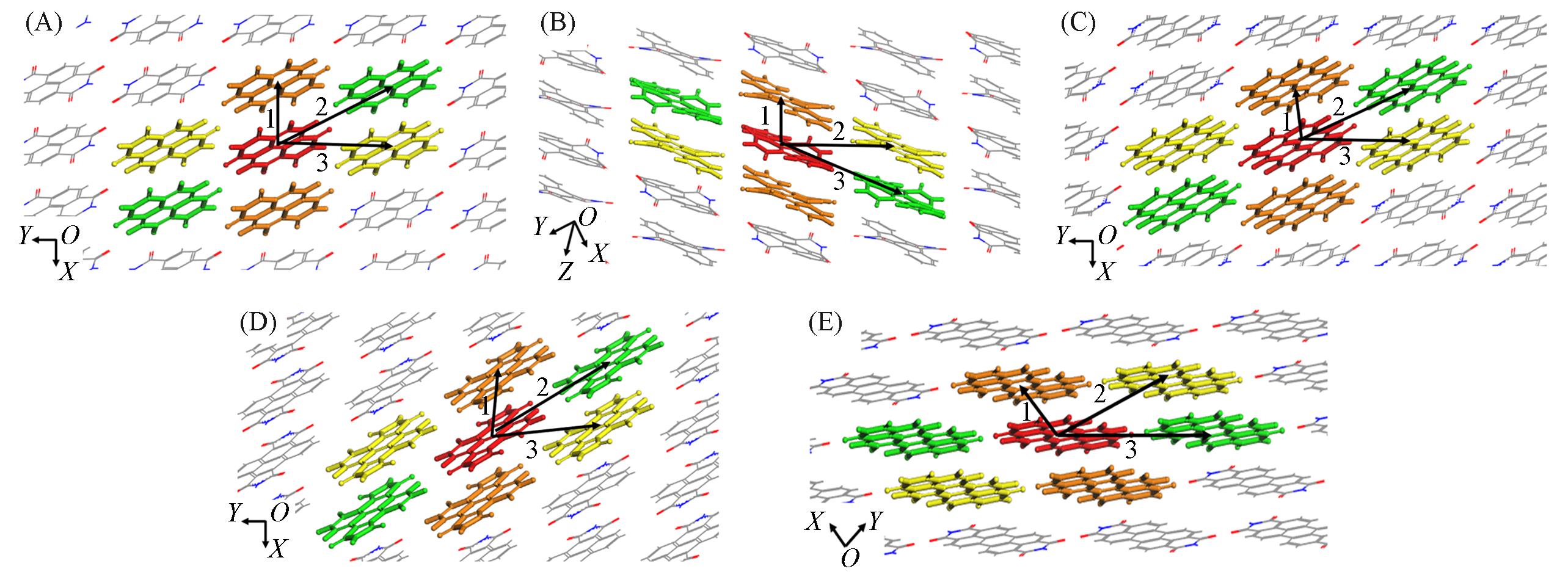
| Path | A1 | A2 | A3 | A3** | A4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | |
| 1 | 4.823 | 65.6 | 30.7 | 5.037 | 91.8 | 58.7 | 4.792 | 65.1 | 63.0 | 4.795 | 26.2 | 7.3 | 4.730 | 33.0 | 113.2 |
| 2 | 8.304 | 28.4 | 8.3 | 10.034 | 5.3 | 4.2 | 9.034 | 10.3 | 36.8 | 8.164 | 8.7 | 36.1 | 9.272 | 41.3 | 2.8 |
| 3 | 9.383 | 24.3 | 1.1 | 11.314 | 4.6 | 6.3 | 9.710 | 6.5 | 5.8 | 10.458 | 10.9 | 1.9 | 11.997 | 10.5 | 0.3 |
Table 4 Electron/hole transfer integrals(Ve /Vh) for the studied molecules, and the centre-of-mass distances(d) between neighbouring dimers
| Path | A1 | A2 | A3 | A3** | A4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | d/nm | Ve/meV | Vh/meV | |
| 1 | 4.823 | 65.6 | 30.7 | 5.037 | 91.8 | 58.7 | 4.792 | 65.1 | 63.0 | 4.795 | 26.2 | 7.3 | 4.730 | 33.0 | 113.2 |
| 2 | 8.304 | 28.4 | 8.3 | 10.034 | 5.3 | 4.2 | 9.034 | 10.3 | 36.8 | 8.164 | 8.7 | 36.1 | 9.272 | 41.3 | 2.8 |
| 3 | 9.383 | 24.3 | 1.1 | 11.314 | 4.6 | 6.3 | 9.710 | 6.5 | 5.8 | 10.458 | 10.9 | 1.9 | 11.997 | 10.5 | 0.3 |
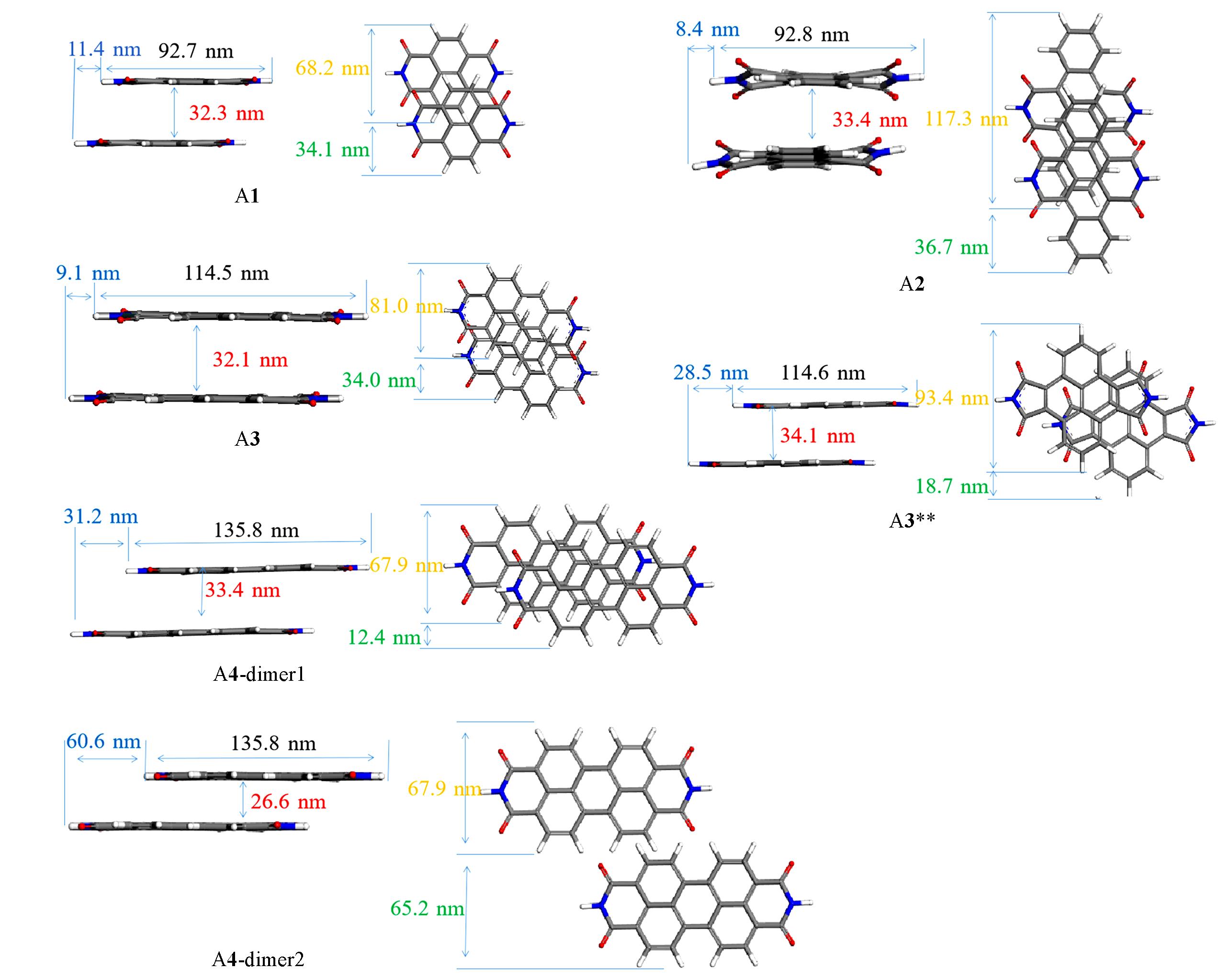
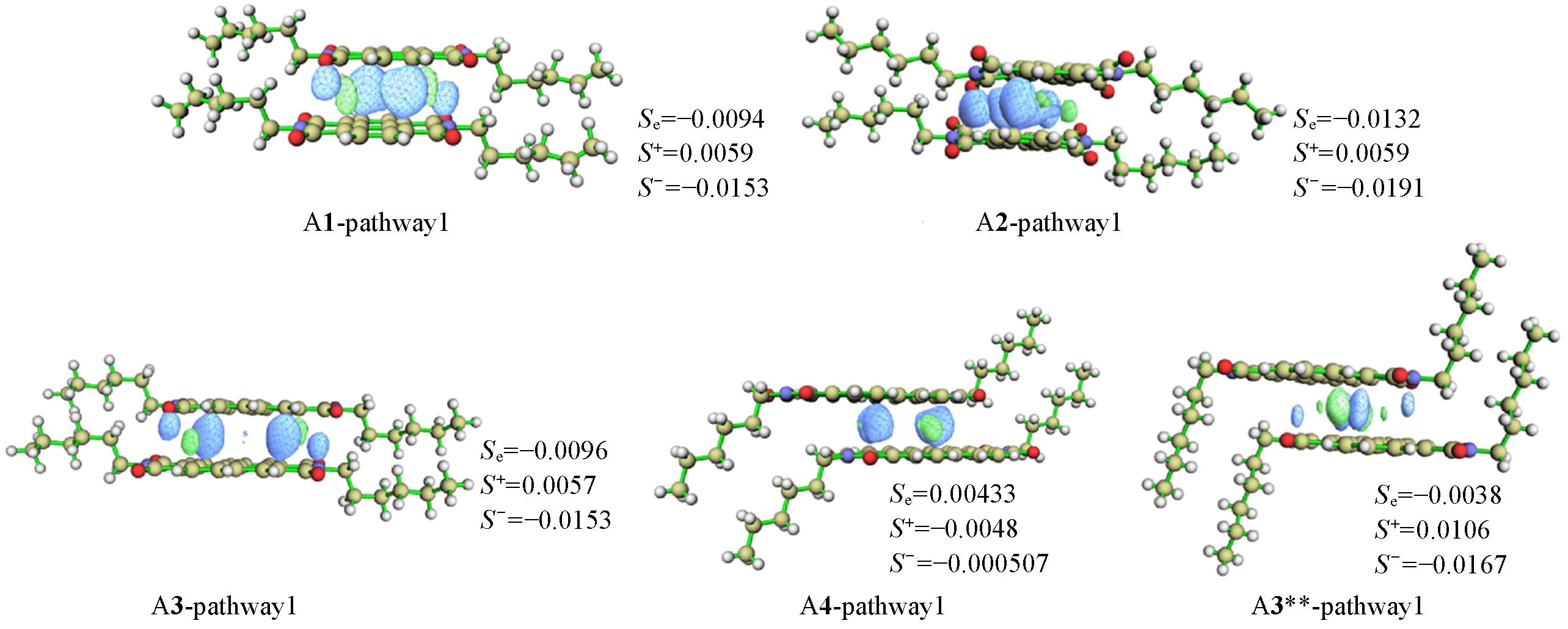
| Molecule | Eelec/(kJ·mol-1) | Eind/(kJ·mol-1) | Edisp/(kJ·mol-1) | Eexch/(kJ·mol-1) | Eattraction/Erepulsion | ESCS-SAPT0/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| A1 | -46.94(16.55%) | -13.35(4.70%) | -132.22(46.60%) | 91.17(32.15%) | 2.11 | -101.30 |
| A2 | -81.13(16.63%) | -20.21(4.15%) | -224.56(46.04%) | 161.80(33.18%) | 2.01 | -164.10 |
| A3 | -59.29(15.34%) | -15.94(4.12%) | -184.68(47.80%) | 126.40(32.71%) | 2.05 | -133.51 |
| A3** | -45.48(12.51%) | -12.68(3.48%) | -193.93(53.29%) | 111.76(30.71%) | 2.26 | -140.33 |
| A4 | -60.38(12.32%) | -16.74(3.42%) | -251.84(51.40%) | 161.04(32.86%) | 2.04 | -167.91 |
Table 5 Calculated intermolecular interaction energy of the nearest dimer for molecules A1—A4 and its corresponding energy decompositions by SAPT0
| Molecule | Eelec/(kJ·mol-1) | Eind/(kJ·mol-1) | Edisp/(kJ·mol-1) | Eexch/(kJ·mol-1) | Eattraction/Erepulsion | ESCS-SAPT0/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| A1 | -46.94(16.55%) | -13.35(4.70%) | -132.22(46.60%) | 91.17(32.15%) | 2.11 | -101.30 |
| A2 | -81.13(16.63%) | -20.21(4.15%) | -224.56(46.04%) | 161.80(33.18%) | 2.01 | -164.10 |
| A3 | -59.29(15.34%) | -15.94(4.12%) | -184.68(47.80%) | 126.40(32.71%) | 2.05 | -133.51 |
| A3** | -45.48(12.51%) | -12.68(3.48%) | -193.93(53.29%) | 111.76(30.71%) | 2.26 | -140.33 |
| A4 | -60.38(12.32%) | -16.74(3.42%) | -251.84(51.40%) | 161.04(32.86%) | 2.04 | -167.91 |
| Molecule | μ1Dmax/(cm2·V‒1·s‒1) | μ2D/(cm2·V‒1·s‒1) | μexp/(cm2·V‒1·s‒1) |
|---|---|---|---|
| A1 | 0.19 | 0.07 | 0.70[ |
| A2 | 0.96 | 0.02 | 0.03[ |
| A3 | 0.32 | 0.06 | — |
| A3** | 0.10 | 0.03 | 0.51[ |
| A4 | 0.71 | 0.15 | — |
Table 6 Simulated electron mobilities of all compounds investigated and available experimental values*
| Molecule | μ1Dmax/(cm2·V‒1·s‒1) | μ2D/(cm2·V‒1·s‒1) | μexp/(cm2·V‒1·s‒1) |
|---|---|---|---|
| A1 | 0.19 | 0.07 | 0.70[ |
| A2 | 0.96 | 0.02 | 0.03[ |
| A3 | 0.32 | 0.06 | — |
| A3** | 0.10 | 0.03 | 0.51[ |
| A4 | 0.71 | 0.15 | — |
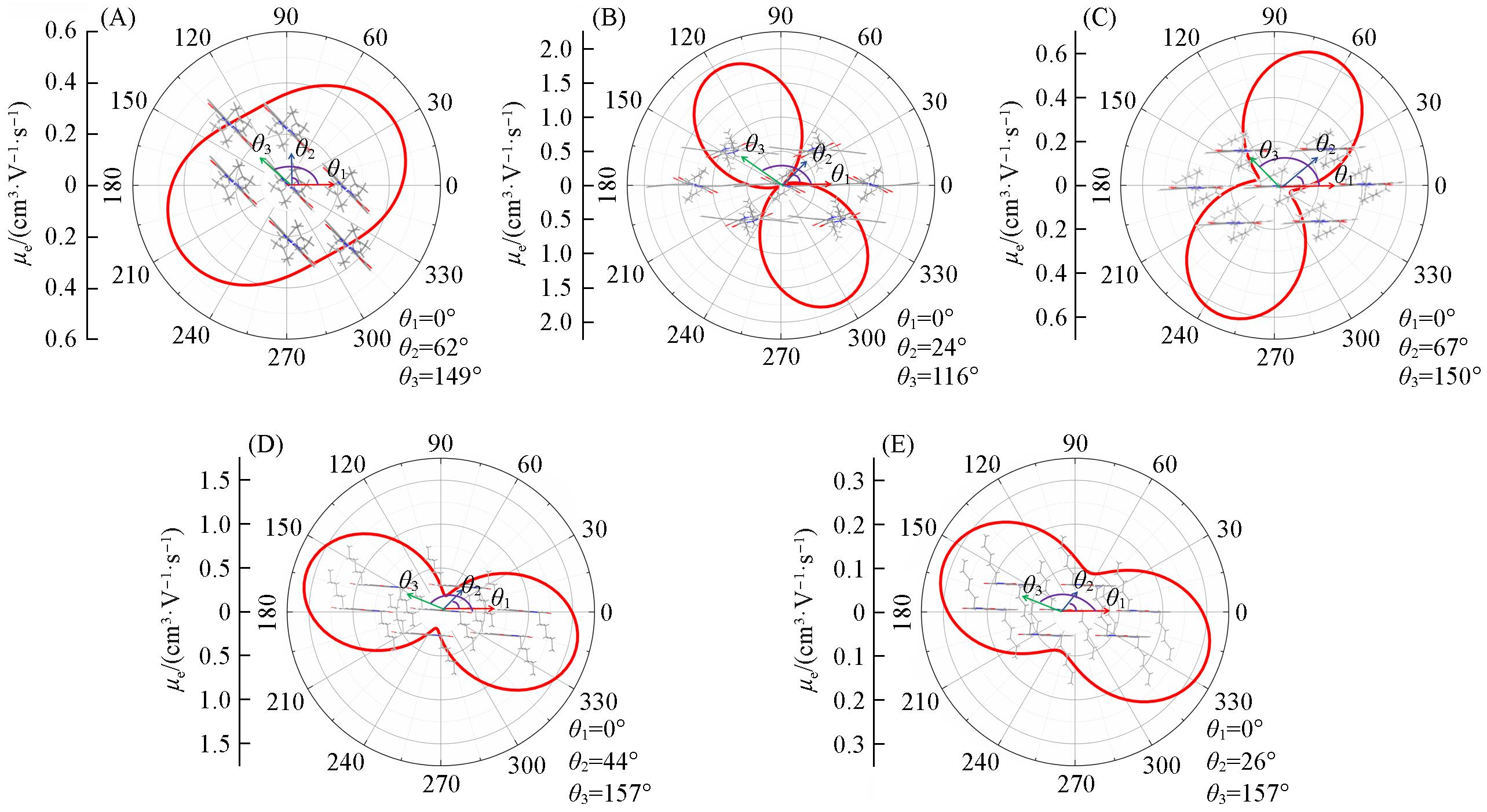
| 1 | Yun H. J., Lee G. B., Chung D. S., Kim Y. H., Kwon S. K., Adv. Mater., 2014, 26(38), 6612—6616 |
| 2 | Dou L., Liu Y., Hong Z., Li G., Yang Y., Chem. Rev., 2015, 115(23), 12633—12665 |
| 3 | Panigrahi D., Hayakawa R., Wakayama Y., Adv. Funct. Mater., 2023, 33(20), 2213899 |
| 4 | Liu X., Anderson C. L., Liu Y., Acc. Chem. Res., 2023, 56(12), 1669—1682 |
| 5 | Zhang T., Xiao Y., Wang H., Kong S., Huang R., Au V. K. M., Yu T., Huang W., Angew. Chem. Int. Ed., 2023, e202301896 |
| 6 | Prache O., Displays., 2001, 22(2), 49—56 |
| 7 | Lee S. Y., Yasuda T., Yang Y. S., Zhang Q. S., Adachi C., Angew. Chem. Int. Ed., 2014, 53(25), 6402—6406 |
| 8 | Zhang H., Wang C., Li X., Jing J., Sun Y., Liu Y., Sol. Energy, 2017, 157(15), 71—80 |
| 9 | Anthopoulos T. D., Setayesh S., Smits E., Clle M., Leeuw D. M. D., Adv. Mater., 2010, 18(14), 1900—1904 |
| 10 | Jedaa A., Burkhardt M., Zschieschang U., Klauk H., Habich D., Schmid G., Halik M., Org. Electronics, 2009, 10(8), 1442—1447 |
| 11 | Tang M. L., Reichardt A. D., Wei P., Bao Z., J. Am. Chem. Soc., 2009, 131(14), 5264—5273 |
| 12 | Haddon R. C., Chi X., Itkis M. E., Anthony J. E., Eaton D. L., Siegrist T., Mattheus C. C., Palstra T. T. M., J. Phys. Chem. B, 2002, 106(33), 8288—8292 |
| 13 | Jones B. A., Facchetti A., Wasielewski M. R., Marks T. J., J. Am. Chem. Soc., 2007, 129(49), 15259—15278 |
| 14 | Dou J. H., Zheng Y. Q., Yao Z. F., Yu Z. A., Lei T., Shen X., Luo X. Y., Sun J., Zhang S. D., Ding Y. F., Han G., Yi Y., Wang J. Y., Pei J., J. Am. Chem. Soc., 2015, 137(50), 15947—15956 |
| 15 | Bhosale S. V., Jani C. H., Langford S. J., Chem. Soc. Rev., 2008, 37(2), 331—342 |
| 16 | Sakai N., Mareda J., Vauthey E., Matile S., Chem. Commun., 2010, 46(24), 4225—4237 |
| 17 | Jia T., Li Z., Ying L., Jia J., Fan B., Zhong W., Pan F., He P., Chen J., Huang F., Cao Y., Macromol. Rapid. Comm., 2018, 39(14), 1700765 |
| 18 | Singh T. B., Erten S., Günes S., Zafer C., Turkmen G., Kuban B., Teoman Y., Sariciftci N. S., Icli S., Org. Electron., 2006, 7(6), 480—489 |
| 19 | Shukla D., Nelson S. F., Freeman D. C., Rajeswaran M., Ahearn W. G., Meyer D. M., Carey J. T., Chem. Mater., 2008, 20(24), 7486—7491 |
| 20 | Katsuta S., Tanaka K., Maruya Y., Mori S., Masuo S., Okujima T., Uno H., Nakayama K. I., Yamada H., Chem. Mater., 2011, 47(36), 10112—10114 |
| 21 | Wu Z. H., Huang Z. T., Guo R. X., Sun C. L., Chen L. C., Sun B., Shi Z. F., Shao X., Li H., Zhang H. L., Angew. Chem. Int. Ed., 2017, 56(42), 13031—13035 |
| 22 | Li Y., Yao Z., Xie J., Han H., Yang G., Bai X., Pei J., Zhao D., J. Mater. Chem. C, 2021, 9(24), 7599—7606 |
| 23 | Pitchaimani J., Kundu A., Anthony S. P., Moon D., Madhu V., Chem. Select., 2020, 5(6), 2070—2074 |
| 24 | Cheng Y. C., Silbey R. J., da Silva Filho D. A., Calbert J. P., Cornil J., Brédas J. L., J. Mater. Chem. C, 2003, 118(8), 3764—3774 |
| 25 | Hutchison G. R., Ratner M. A., Marks T. J., J. Am. Chem. Soc., 2005, 127(7), 2339—2350 |
| 26 | Deng W. Q., Goddard W. A., J. Phys. Chem. B, 2004, 108(25), 8614—8621 |
| 27 | Marcus R. A., Rev. Mod. Phys., 1993, 65(3), 599—610 |
| 28 | Zhang S. F., Chen X. K., Fan J. X., Ren A. M., Org. Electron., 2013, 14(2), 607—620 |
| 29 | Schein L. B., Mcghie A. R., Phys. Rev. B, 1979, 20(4), 1631—1639 |
| 30 | Brédas J. L., Beljonne D., Coropceanu V., Cornil J., Chem. Rev., 2004, 104(11), 4971—5004 |
| 31 | Deng W. Q., Sun L., Huang J. D., Chai S., Wen S. H., Han K. L., Nat. Protoc., 2015, 10(4), 632—642 |
| 32 | Rappe A. K., Casewit C. J., Colwell K. S., Goddard W. A. III, Skiff W. M., J. Am. Chem. Soc., 1992, 114(25), 10024—10035 |
| 33 | Maseras F., Morokuma K., J. Comuput. Chem., 1995, 16(9), 1170—1179 |
| 34 | Vreven T., Byun K. S., Komáromi I., Dapprich S., Montgomery John A., Morokuma K., Frisch M. J., J. Chem. Theory Comput., 2006, 2(3), 815 |
| 35 | Chang Y. C., Kuo M. Y., Chen C. P., Lu H. F., Chao I., J. Phys. Chem. C, 2010, 114(26), 11595—11601 |
| 36 | Lin P. P., Zhang S. F., Zhang N. X., Fan J. X., Ji L. F., Guo J. F., Ren A. M., Phys. Chem. Chem. Phys., 2019, 21(6), 3044—3058 |
| 37 | Delgado M. C. R., Kim E. G., Filho D. A. d. S., Bredas J. L., J. Am. Chem. Soc., 2010, 132(10), 3375—3387 |
| 38 | Parker T. M., Burns L. A., Parrish R. M., Ryno A. G., Sherrill C. D., J. Phys. Chem. C, 2014, 140(9), 094106 |
| 39 | Turney J. M., Simmonett A. C., Parrish R. M., Hohenstein E. G., Evangelista F. A., Fermann J. T., Mintz B. J., Burns L. A., Wilke J. J., Abrams M. L., Russ N. J., Leininger M. L., Janssen C. L., Seidl E. T., Allen W. D., Schaefer H. F., King R. A., Valeev E. F., Sherrill C. D., Crawford T. D., Wires. Comput. Mol. Sci., 2012, 2(4), 556—565 |
| [1] | SONG Xin, GAO Shenzheng, XU Shanlei, XU Hao, ZHOU Xinjie, ZHU Mengbing, HAO Rulin, ZHU Weiguo. Progress on the Efficiency Regulation of Organic Solar Cells by Volatile Solid Additives [J]. Chem. J. Chinese Universities, 2023, 44(9): 20230151. |
| [2] | CHEN Hongru, BAI Yang, ZHOU Qiuju, ZHANG Zhiguo. Application of 2D NMR in Organic Photovoltaics [J]. Chem. J. Chinese Universities, 2023, 44(7): 20230104. |
| [3] | WANG Yingjie, PAN Zehui, TAO Zhengyu, WANG Yan, TONG Bihai, FUNG Mankeung, SONG Mingxing. Effect of Intermolecular Interaction on the Properties of Benzophenone-based AIDF Materials [J]. Chem. J. Chinese Universities, 2023, 44(4): 20220562. |
| [4] | WANG Jianqiao, MA Yuguang. Extent and Changeable Rule of HOMO and LUMO Energy of Organic Semiconductors in Nonequilibrium States and a Phenomenological Understanding for the Formation of “Hot Excitons” in OLED [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210856. |
| [5] | LIN Chengce, PENG Boyu, LI Hanying. Recent Progress in Organic Single Crystal Integrated Circuits [J]. Chem. J. Chinese Universities, 2021, 42(6): 1672. |
| [6] | MA Zhuoyuan, WANG Dayang. Status and Prospect of Surface Wettability of Molecular Self-assembled Monolayers [J]. Chem. J. Chinese Universities, 2021, 42(4): 1031. |
| [7] | ZHU Lei,HAN Junyan,CHANG Haizhen,QIU Yuyuan,ZHANG Yanan,PENG Danni,HU Wei,MIAO Shaobin. Different Pathways for the Cyclocondensation Reactions of 1,2-Diamine and 1,2-Diketone† [J]. Chem. J. Chinese Universities, 2018, 39(12): 2686. |
| [8] | CHEN Jiuju. Theoretical Studies on the of Ambipolar Charge Transport in Terazulene Single Crystal† [J]. Chem. J. Chinese Universities, 2016, 37(1): 121. |
| [9] | SI Pengfei, LUO Faliang, HAI Mei. Intermolecular Interactions and Crystallization and Melting Behavior of Poly(L-lactic acid)/4,4'-Thiobis Phenol Blends† [J]. Chem. J. Chinese Universities, 2015, 36(1): 188. |
| [10] | YUAN Wei, REN Qingjiang, SUN Hengda, LI Hui, CHENG Yanxiang, MA Dongge. Effect of Peripheral Substituents on Luminescent Properties of the Porphyrin Platinum(Ⅱ) Complexes† [J]. Chem. J. Chinese Universities, 2014, 35(6): 1229. |
| [11] | MENG Su-Ci, YIN Xiu-Lian, MA Jing, XIE Ji-Min. Theoretical Studies on Solvent Effects and Intermolecular Interactions of Organic π-Conjugated Ligand in Solutions [J]. Chem. J. Chinese Universities, 2012, 33(11): 2492. |
| [12] | LIANG Xue, SUN Tao, WANG Yi-Bo. Symmetry-adapted Perturbation Theory Study on the Nature of Benzene-halogen(X2, X=F, Cl, Br, I) [J]. Chem. J. Chinese Universities, 2012, 33(03): 541. |
| [13] | ZHAO Jian-Xin1, QIAO Yi-Tao1, FENG Jing1, LUO Zhao-Feng2, YUAN Zhi1*. Interaction of Copolymer-Zn with Polypeptide [J]. Chem. J. Chinese Universities, 2008, 29(3): 658. |
| [14] | WANG Zhao-Xu, ZHANG Jing-Chang, CAO Wei-liang. Theoretical Study on Intermolecular Interactions BetweenHCN(HNC) and NH3, H2O, HF [J]. Chem. J. Chinese Universities, 2007, 28(2): 320. |
| [15] |
TIAN Hong-Kun1,2, SHI Jian-Wu1,2, YAN Dong-Hang1, WANG Li-Xiang1, GENG Yan-Hou1, WANG Fo-Song1 . Synthesis and Characterization of 2,5-Bis(2-phenanthrenyl)-thieno[3,2-b]thiophene and Its Application in Organic Thin-film Transistors [J]. Chem. J. Chinese Universities, 2006, 27(9): 1677. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||