



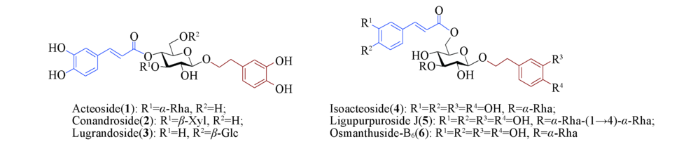
苯丙素苷(PPGs)是一类含有C6—C3芳香核片段的天然糖苷类化合物, 广泛存在于植物中. 对该类糖苷的研究始于上世纪60年代, 目前已有相当数量并具有各类生理活性的苯丙素苷被分离鉴定[1,2,3,4,5,6,7,8]. 苯丙素苷的核心糖单元是β-D-吡喃葡萄糖, 其糖环上不同位置羟基可以被不同基团修饰: 异头位羟基常连接取代苯乙基作为苷元; 2-, 4-或6-位羟基可连接反式取代肉桂酰基; 糖环的2-, 3-, 4-或6-位羟基常连接有L-鼠李糖(Rha)、D-半乳糖(Gal)、D-木糖(Xyl)和D-葡萄糖(Glc)等单糖或二糖, 形成结构更为复杂的苯丙素苷(Scheme 1). 活性研究结果表明, 苯丙素苷具有抗菌消炎[9]、抗肿瘤[10,11,12]、抗病毒[13]、抗氧化[14,15,16]及保肝护肝[17,18]等生物活性. 由于该类化合物在植物体内含量较低且分离困难, 难以满足对其生物和药理活性进行深入研究的需求.
Scheme 1
20世纪90年代起, 有关苯丙素苷的合成研究工作不断被报道. 1998年, Cai等[19]合成了Osmanthuside-B6(6)的2-O-乙酰基衍生物; 1999年, Kawada等[20]和van Boom等[21]先后报道了Acteoside(1)的全合成; 2000年和2002年, Kawada等[22,23]先后报道了Conandroside(2)和Isoacteoside(4)的全合成. 上述工作使用了较多的保护基操作, 而且部分保护基与苯丙素苷具有的α,β不饱和酯结构不相容, 导致合成效率不高. 为降低苯丙素苷类分子的合成路线对保护基的依赖, 缩减合成步骤并提高效率, Judeh等[24]在2-氨基乙基二苯基硼酸酯活化下, 用卤苷给体与多羟基受体进行区域选择性糖苷化, 生成相应的原酸酯, 乙酰基保护受体的剩余羟基后, 在三氟甲磺酸三甲基硅酯(TMSOTf)促进下转化为1→3连接的二糖, 进而完成苯丙素苷Osmanthuside-B6(6)的全合成.
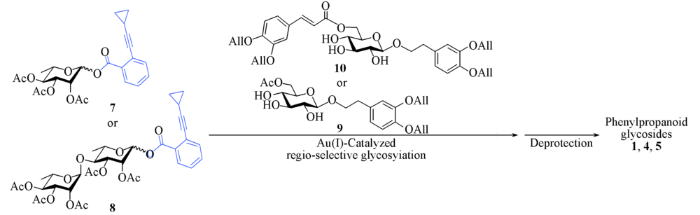
2008年以来, 本课题组[25,26,27,28,29,30]发展了以糖基邻炔基苯甲酸酯为给体的金(Ⅰ)催化糖苷化反应, 该反应的活化机制与经典的糖苷化反应不同, 反应条件中性温和, 已被应用于多个天然产物的高效合成[31,32,33,34,35]. 利用葡萄糖硫苷中各个羟基活性的差异, 以该方法进行区域选择性糖苷化, 可以得到关键的α-(1→3)连接的二糖化合物. 在苯丙素苷的合成中发现, 该二糖硫苷需要转化成其它给体才能与取代苯乙醇进行糖苷化[36]. 基于此, 本文改进了合成路线, 先完成取代苯乙醇葡萄糖苷的合成, 再进行区域选择性糖苷化构建α-(1→3)糖连接, 并完成了天然苯丙素苷毛蕊花糖苷(Acteoside, 1)和异毛蕊花糖苷(Isoacteoside, 4)的合成及紫茎女贞苷J(Ligupurpuroside J, 5)的首次全合成(Scheme 2).
Scheme 2
1 实验部分
1.1 试剂与仪器
N-碘代丁二酰亚胺(NIS, 纯度98%)、四(三苯基膦)钯(纯度99%)和N,N-二异丙基乙胺(DIPEA, 纯度99.5%)购自萨恩化学技术(上海)有限公司; 三氟甲磺酸三甲基硅酯(TMSOTf, 纯度99%)购自艾览(上海)化工科技有限公司; N-甲基咪唑(NMI, 纯度99%)和氯乙酰(纯度98%)购自上海阿拉丁生化科技股份有限公司; 乙酸酐(分析纯)购自国药集团化学试剂有限公司; 二环己基碳二亚胺(DCC, 化学纯)购自上海天莲化工科技有限公司; 4-二甲氨基吡啶(DMAP, 纯度99%)和质量分数30%~33%甲胺的甲醇溶液购自上海百灵威化学技术有限公司; 草酰氯(纯度>98%)购自梯希爱(上海)化成工业发展有限公司; 四丁基氟化铵四氢呋喃溶液(1 mol/L)和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐 (EDCI, 纯度99%)购自上海毕得医药科技有限公司. 常用溶剂采用微波炉活化的0.4 nm分子筛进行干燥; 柱层析所用溶剂均为分析纯.
Agilent-500型核磁共振波谱仪[NMR, 以四甲基硅烷(TMS)作为内标, 美国Agilent公司]; Agilent 6224 TOF LC/MS型和Agilent TOF LC/MS 1260-6230型高分辨质谱仪(HRMS, 美国Agilent公司); Anton Paar MCP 5500型旋光仪(光源为钠源589 nm, 奥地利Anton Paar公司); ZF-Ⅰ型三用紫外分析仪(波长为254 nm, 上海豫康科教仪器设备有限公司).
1.2 实验过程
参照文献方法合成了三苯基膦金(I)双(三氟甲磺酰基)亚胺盐(PPh3AuNTf2)和邻环丙基乙炔基苯甲酸[26]、3,4-二-O-烯丙氧基苯乙基-β-D-吡喃葡萄糖苷(11)[37]、2-环丙乙炔基苯甲酰基2,3,4-三-O-乙酰基-L-吡喃鼠李糖苷(7)[25,38]、三氯乙酰亚胺酯基2,3,4-三-O-乙酰基-L-吡喃鼠李糖苷(12)[39]、3,5-二甲基-4-(2′-苯乙炔苯基)苯基2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖苷(13)[40]、3,4-二-O-烯丙氧基-E-肉桂酸(14)[41]和叔丁基二甲基硅基2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-异亚丙基-α-L-吡喃鼠李糖苷(15)[40].
1.2.1 3,4-二-O-烯丙氧基苯乙基-6-O-乙酰基-β-D-吡喃葡萄糖苷(9)的合成 将4.6 g(11.6 mmol)化合物11分散于250 mL乙酸乙酯中, 加入125 µL(2.3 mmol)浓硫酸, 于40 ℃反应至用薄层色谱(TLC)监测原料基本消耗完全(约40 h); 依次用饱和NaHCO3水溶液和饱和NaCl溶液洗涤, 用乙酸乙酯萃取, 合并有机相, 用无水Na2SO4干燥, 经过滤、浓缩、柱层析[V(二氯甲烷):V(甲醇)=40:1至20:1]得到3.2 g白色固体9, 产率为62%, 回收产率为68%.
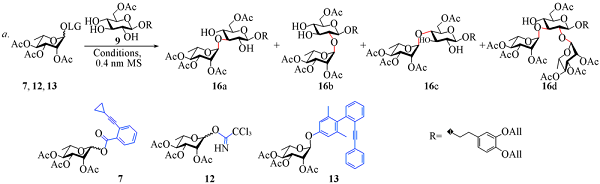
1.2.2 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-乙酰基-β-D-吡喃葡萄糖苷(16a)及具有类似结构的副产物16b, 16c和16d的合成 将137 mg(0.3 mmol)糖苷化给体7和263 mg(0.6 mmol)受体9溶解于13 mL干燥的二氯甲烷中, 加入1 g活化的0.4 nm分子筛, 在氩气保护下于室温搅拌反应0.5 h. 将反应体系温度降至-60 ℃, 加入PPh3AuNTf2的二氯甲烷溶液(0.03 mol/L, 1 mL), 在-60 ℃下反应10 h; 升温至-50 ℃继续反应30 h; 升温至室温并加入三乙胺淬灭反应. 将反应液垫硅藻土过滤并浓缩, 经柱层析[V(石油醚):V(乙酸乙酯)=4:1至2:1至1:1], 得到白色泡沫状固体16a和16d的混合物共125 mg, 其中16a 109 mg, 产率为51%, 16d 15 mg, 产率为10%, 另外得到40 mg白色固体16b(产率为19%)和很少量的16c.
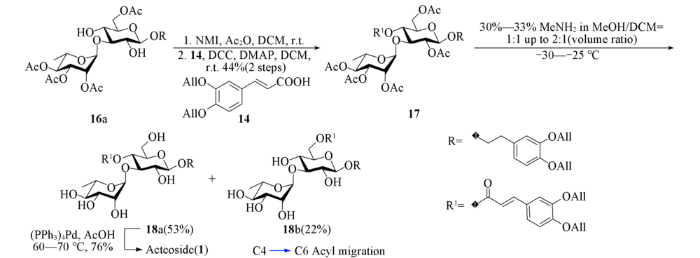
1.2.3 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-2,6-二-O-乙酰基-4-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(17)的合成 将245 mg(0.35 mmol)化合物16a和82 µL(1.0 mmol) NMI溶解于15 mL干燥的二氯甲烷中, 在氩气保护及冰浴下缓慢加入39 µL, 0.4 mmol乙酸酐, 于0 ℃反应24 h; 升温至室温继续反应15 h, 反应液用饱和食盐水洗涤, 用乙酸乙酯萃取水相2次, 合并有机相, 用无水Na2SO4干燥, 经过滤、浓缩及快速柱层析[V(石油醚):V(乙酸乙酯)=2:1至1:1]得2-酰基产物粗品. 将该粗品、182 mg(0.7 mmol)化合物14、63 mg(0.5 mmol)DMAP和180 mg(0.9 mmol)DCC溶解于5 mL干燥的二氯甲烷中, 于室温反应至原料消耗完全; 用乙酸乙酯稀释, 垫硅藻土过滤, 用饱和NaCl溶液洗涤, 有机相用无水Na2SO4干燥, 经过滤、浓缩、柱层析[V(石油醚): V(丙酮)=6:1至4:1]得到153 mg白色泡沫状固体17, 产率为44%.
1.2.4 3,4-二-O-烯丙氧基苯乙基-α-L-吡喃鼠李糖-(1→3)-4-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(18a)的合成 将60 mg(0.06 mmol)化合物17溶解于3 mL 二氯甲烷中, 于-30 ℃加入 3 mL 质量分数为30%~33%甲胺的甲醇溶液, 保持低温反应5 h. 将体系温度升至-25 ℃反应5 h, 浓缩后经柱层析[V(二氯甲烷):V(甲醇)=40:1]得到25 mg化合物18a, 产率为53%.
1.2.5 Acteoside(1)的合成 将24 mg(30 μmol)化合物18a和52 mg(45 μmol) (PPh3)4Pd溶解于3 mL脱气乙酸中, 于60 ℃反应5 h, 将体系温度升至70 ℃反应至原料消耗完全. 浓缩后用制备板([V(二氯甲烷):V(甲醇):V(乙酸)=3:1:0.1)分离后, 经反相C18柱层析[V(水):V(甲醇)=4:1至2:1]得到14 mg棕黄色固体Acteoside, 产率为76%.
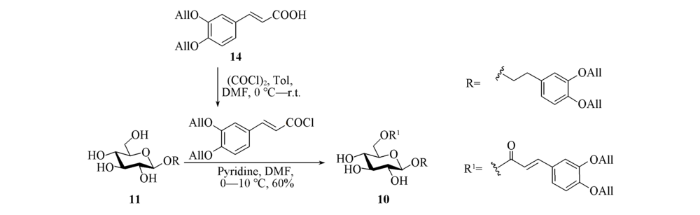
1.2.6 3,4-二-O-烯丙氧基苯乙基-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(10)的合成 将2.6 g(10.0 mmol)化合物14溶解于50 mL甲苯中, 在氩气保护下, 于0 ℃先后加入15 µL N,N-二甲基甲酰胺和6 mL草酰氯; 保持0 ℃反应0.5 h, 将体系温度升至室温继续反应3 h, 浓缩, 抽真空 1 h, 备用. 将3.3 g(8.32 mmol)化合物11溶解于60 mL干燥的二氯甲烷和5 mL干燥的吡啶中, 在氩气保护下, 于-10 ℃通过注射器加入10 mL(1.0 mol/L)新制备的酰氯二氯甲烷溶液; 于-5 ℃反应10 h后升温至10 ℃继续反应10 h; 将体系用乙酸乙酯稀释, 依次用稀盐酸(1 mol/L)和饱和NaCl溶液洗涤, 用无水Na2SO4干燥, 经过滤、浓缩、柱层析[V(石油醚):V(乙酸乙酯)=1:1至1:2至V(二氯甲烷):V(甲醇)=15:1]得到3.0 g白色固体10, 产率为56%, 回收产率为63%.
1.2.7 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(19a)和3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→2)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(19b)的合成 以46 mg (0.1 mmol)给体7和127.7 mg(0.2 mmol)受体10为原料, 参照类似合成化合物16a的选择性糖苷化条件制得化合物19a, 粗品经柱层析[V(石油醚):V(乙酸乙酯)=4:1, 2:1, 1:1]得到50 mg白色固体19a, 产率为55%, 另外得到20 mg 19b, 产率为22%.
1.2.8 3,4-二-O-烯丙氧基苯乙基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(18b)的合成 将45.6 mg(0.05 mmol)化合物19a溶解于5 mL甲醇和1 mL二氯甲烷的混合溶剂中, 于0 ℃加入35 µL(0.5 mmol)乙酰氯, 逐渐升温至室温反应30 h, 用三乙胺淬灭反应, 浓缩后经柱层析[V(二氯甲烷):V(甲醇)=30:1]得到29.4 mg化合物18b, 产率为75%. 另外, 以36 mg(40 µmol)化合物19a为原料, 使用甲胺条件脱除乙酰基, 参照类似合成化合物18a的方法得到29 mg化合物18b, 产率为92%.
1.2.9 Isoacteoside(4)的合成 以20 mg(25 µmol)化合物18b为原料, 参照类似18a脱除烯丙基的方法得到12 mg棕黄色固体Isoacteoside, 产率为79%.
1.2.10 2-环丙乙炔基苯甲酰基- 2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-L-吡喃鼠李糖苷(8)的合成 将295 mg(0.5 mmol)化合物15加入10 mL乙酸水溶液(体积分数为80%)中, 于50 ℃反应4 h, 用乙酸乙酯稀释并用饱和NaHCO3溶液中和, 用乙酸乙酯萃取2次, 有机相用无水Na2SO4干燥, 过滤, 浓缩后与甲苯共沸除水2次得到浅黄色固体; 将该固体和61 mg(0.5 mmol)DMAP溶于 5 mL干燥的吡啶中, 于0 ℃通过注射器加入0.2 mL(2.0 mmol)乙酸酐; 逐渐升温至室温并反应至原料消耗完全(约3 h, TLC监测); 加入1 mL甲醇于室温搅拌1 h以消耗多余的乙酸酐, 除去大部分溶剂, 用乙酸乙酯稀释并依次用稀盐酸(1 mol/L)和饱和NaHCO3溶液洗涤, 经无水Na2SO4干燥、过滤、浓缩得到粗品; 将粗品溶于10 mL四氢呋喃中, 冰浴下滴加1 mol/L四丁基氟化铵四氢呋喃溶液, 冰浴下反应0.5 h, 升温至室温并反应至原料消耗完全; 用乙酸乙酯稀释, 饱和NaCl溶液洗涤, 用乙酸乙酯萃取 2次, 有机相用无水Na2SO4干燥, 过滤, 浓缩后与甲苯共沸除水2次, 得到异头位裸露的二糖粗品; 将该粗品、121 mg(0.65 mmol)邻环丙基乙炔基苯甲酸, 124 mg(0.65 mmol)EDCI, 79 mg(0.65 mmol)DMAP和176 µL(1.0 mmol)DIPEA溶解于5 mL干燥的二氯甲烷中, 室温下反应至原料消耗完全, 用乙酸乙酯稀释, 有机相用饱和NaCl溶液洗涤, 经无水Na2SO4干燥、过滤、浓缩、柱层析[V(石油醚):V(乙酸乙酯)=3:1]得到244 mg白色泡沫状固体8, 产率为71%.
1.2.11 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(20a)和3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-α-L-吡喃鼠李糖-(1→2)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(20b)的合成 以34.4 mg(0.05 mmol)给体8和63.9 mg(0.1 mmol)受体10为原料, 参照类似合成化合物16a的选择性糖苷化条件制得化合物20, 粗品经柱层析[V(石油醚):V(乙酸乙酯)=4:1, 2:1, 1:1]得到32 mg白色固体化合物20a, 产率为57%, 另外得到12 mg化合物20b, 产率为19%.
1.2.12 Ligupurpuroside J(5)的合成 以23 mg(20 µmol)化合物20为原料, 参照类似化合物17脱除乙酰基的方法采用甲胺条件脱除乙酰基, 得到粗品后, 参照类似化合物18a脱除烯丙基的方法得到 9.9 mg棕黄色固体Ligupurpuroside J, 产率为64%.
2 结果与讨论
2.1 Acteoside的全合成
Scheme 3

使用鼠李糖邻炔基苯甲酸酯给体7[25,38]和受体9在Ph3PAuNTf2催化下进行了区域选择性糖苷化反应的尝试(见表1). 首先, 反应在-70 ℃下进行, 使用10%(摩尔分数)的 PPh3AuNTf2作催化剂, 二氯甲烷作溶剂, 反应12 h后以13%的产率得到主要的α-(1→3)糖苷化产物16a, 同时回收了65%的给体 (表1中Entry 1). 为提高给体的转化率, 升高反应温度至-50 ℃并延长反应时间, 以51%的收率得到糖苷化产物16a, 同时获得少部分的α-(1→2)连接的二糖16b(19%)和三糖16d(10%)(表1中Entry 2). 为了缩短反应时间, 进一步升高反应温度至-30 ℃, 发现选择性明显下降(表1中Entry 3). 改变溶剂为甲苯或乙醚, 糖苷化选择性发生逆转, α-(1→2)糖苷化产物16a成为优势产物(表1中Entries 4和5). 将金催化剂替换为新制的PPh3AuOTf, 发现糖苷化选择性同样下降(表1中Entry 6). 为与其它糖苷化方法作对比, 将金(I)催化条件替换为TMSOTf或TMSOTf/N-NIS活化条件, 使用三氯乙酰亚胺酯给体12[39]和3,5-二甲基-4-(2′-苯乙炔苯基)苯基(EPP)糖苷给体13[40]分别与受体9在-50 ℃下进行反应, 反应的选择性明显降低, 同时给体7的选择性也几乎消失(表1中Entries 7~9). 上述结果证明, Au(I)催化糖苷化反应的条件温和, 在低温下可以实现一定的区域选择性. 产物中二糖16a和少量的三糖16d极性相似, 难以分离, 但这并不影响后续反应, 再经过2步反应后即可分离除去相应杂质.
Table 1 Regioselective glycosylation of acceptor 9 with donors 7, 12 and 13a
| Entry | Donor | Catalyst | Amount(%, mass fraction) | Solvent | Temp./℃ | Product | Yield(%) or molar ratio |
|---|---|---|---|---|---|---|---|
| 1 | 7 | PPh3AuNTf2 | 10 | DCM | -70 | 16a | 13b |
| 2 | 7 | PPh3AuNTf2 | 10 | DCM | -50 | 16a, 16b, 16c, 16d | 51b, 19b, Trace, 10b |
| 3 | 7 | PPh3AuNTf2 | 10 | DCM | -30 | 16a, 16b | 16a/16b=1.5—2:1c |
| 4 | 7 | PPh3AuNTf2 | 10 | Toluene | -50 | 16a, 16b | 16a/16b=1:1.5—2c |
| 5 | 7 | PPh3AuNTf2 | 10 | Ether | -50 | 16a, 16b | 16a/16b=1:1.5—2c |
| 6 | 7 | PPh3AuOTf | 10 | DCM | -50 | 16a, 16b | 16a/16b=1.2—1.5:1c |
| 7 | 12 | TMSOTf | 5—20 | DCM | -50 | 16a, 16b, 16c, 16d | 29d, 24d, 10d, 7d |
| 8 | 13 | TMSOTf/NIS | 120/20 | DCM | -50 | 16a, 16b, 16c, 16d | 28d, 21d, 14d, 6d |
| 9 | 7 | TMSOTf/NIS | 120/10 | DCM | -50 | 16a, 16b, 16c, 16d | 26d, 22d, 8d, 28d |
b. Isolated yield; c. the ratio was estimated by TLC; d. NMR yield.
经区域选择性糖苷化反应得到二糖16a后, 对葡萄糖2-位进行了选择性保护. 由于该位点羟基位阻较大, 硅基和氯乙酰基均很难对其进行保护, 最后采用体积较小的乙酰基进行保护. 通过筛选反应条件发现, 使用乙酸酐作为乙酰化试剂, NMI作为碱, 室温下可以相对较高的转化率和产率得到2-乙酰化产物, 但其氢谱包含2套信号(比例约为5:1), 推测杂质可能为4-乙酰化产物, 两者极性一致, 用柱层析无法分离; 该粗品与烯丙基保护的咖啡酸片段14[41]缩合后得到的化合物17可分离纯化, 2步反应的产率为44%.
随后进行了保护基的脱除. 计划先脱除烯丙基后脱除乙酰基, 但实验中发现在甲胺条件下脱除乙酰基时会产生较多副产物, 并且与终产物极性相似, 可以是咖啡酸支链断裂的产物, 使用反相C18柱层析分离困难. 因此, 尝试先乙酰基后烯丙基的脱除顺序, 在乙酰基的脱除中, 起初在反应体系中观察 2个主点. 因为葡萄糖2-位位阻较大, 保护基较难脱除, 假设极性较小的产物为葡萄糖2-位乙酰基未脱除产物, 补加甲胺溶液使之向极性较大的产物转化, 但通过核磁共振氢谱分析发现, 这2种产物均无乙酰基的信号, 极性较小的化合物为正常脱乙酰基产物18a, 同时反应过程中部分底物发生了C4→C6酰基迁移, 得到了化合物18b. 该化合物脱除烯丙基后可得到另一种苯丙素苷天然产物Isoacteoside. 类似条件下的酰基迁移现象已有相关文献报道[23]. 最后, 通过调整甲胺溶液与二氯甲烷的比例以及反应温度, 以53%的收率得到产物18a. 化合物18a在四(三苯基膦)钯[(PPh3)4Pd]条件下脱除烯丙基, 以76%的产率得到目标分子Acteoside(见Scheme 4).
Scheme 4
2.2 Isoacteoside和Ligupurpuroside J的合成
Scheme 5
尝试使用鼠李糖给体7与受体10在优化的条件下进行了区域选择性糖苷化反应. 使用2倍摩尔量受体时, 以55%的收率得到α-(1→3)糖苷化产物19a, 同时分离得到22%的α-(1→2)二糖19b. 随后, 对化合物19a进行保护基的脱除. 脱除乙酰基时, 首先尝试了乙酰氯/甲醇条件, 以75%的产率得到化合物18b; 其次, 尝试采用甲胺条件, 保持反应温度在-25~-20 ℃, 则可以92%的产率高效脱除全部乙酰基. 最后, 将化合物18a脱除烯丙基, 以79%的产率顺利得到天然产物Isoacteoside [Scheme 6(A)].
Scheme 6
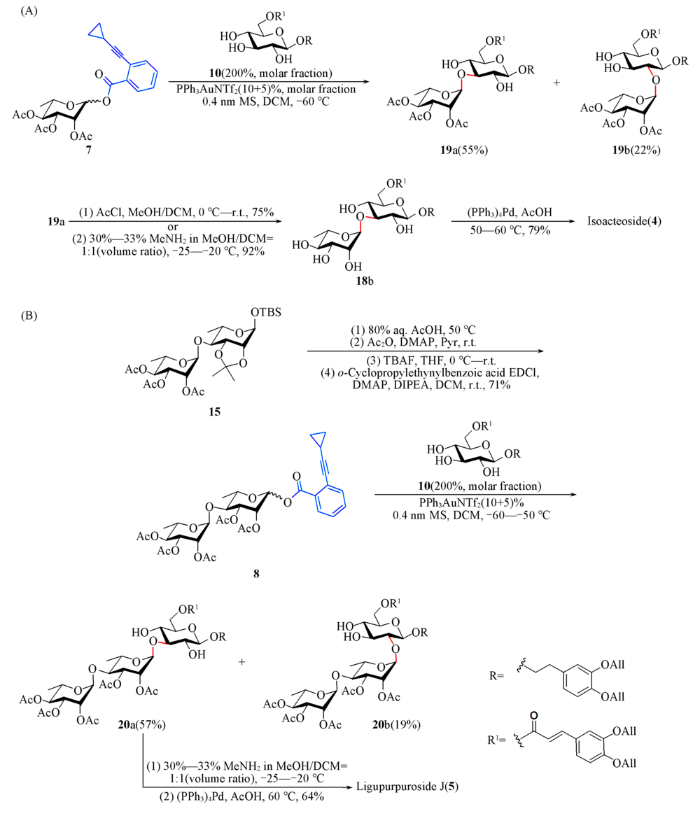
Table 2 Optical rotation values and HRMS data of compounds 8—10, 16—20, Acteoside(1), Isoacteoside(4) and Ligupurpuroside J(5)
| Compd. | [α]D25(c, g/100 mL) | HRMS(calcd.), m/z [M+Na]+ |
|---|---|---|
| 8α | -9.2(c 0.37, CHCl3) | 711.2264(711.2259) |
| 8β | -87.5(c 0.69, CHCl3) | 711.2262(711.2259) |
| 9 | -33.3(c 1.0, CHCl3) | 461.1785(461.1782) |
| 10 | -55.7(c 1.0, CHCl3) | 661.26070(661.26193) |
| 16a | -44.3(c 0.59, CHCl3) | 733.27144(733.26781) |
| 16b | -49.5(c 0.64, CHCl3) | 733.2685(733.2678) |
| 16c | -25.5(c 0.73, CHCl3) | 733.2680(733.2678) |
| 16d | -29.4(c 1.0, CHCl3) | 1005.3580(1005.3574) |
| 17 | -63.2(c 0.68, CHCl3) | 1017.3727(1017.3727) |
| 18a | -52.6(c 0.25, CHCl3) | 807.3201(807.3198) |
| 18b | -36.4(c 0.71, CHCl3) | 807.3200(807.3198) |
| 19a | -40.8(c 0.55, CHCl3) | 933.34910(933.35154) |
| 19b | -81.1(c 1.0, CHCl3) | 933.3519(933.3515) |
| 20a | -39.4(c 1.0, CHCl3) | 1163.4310(1163.4306) |
| 20b | -76.4(c 0.52, CHCl3) | 1163.4311(1163.4306) |
| Acteoside(1) | -69.7(c 0.20, CH3OH) | 647.1960(647.1946) |
| Isoacteoside(4) | -52.1(c 0.20, CH3OH) | 647.1954(647.1946) |
| Ligupurpuroside J(5) | -84.6(c 0.15, CH3OH) | 793.2530(793.2526) |
Table 3 1H and13C NMR data of compounds 8—10, 16—20, Acteoside(1), Isoacteoside(4) and Ligupurpuroside J(5)
| Compd. | 1H NMR (500 MHz), δ | 13C NMR (126 MHz), δ |
|---|---|---|
| 8αa | 7.79 (ddd, J=7.9, 1.4, 0.6 Hz, 1H), 7.46 (dd, J=7.8, 1.4 Hz, 1H), 7.41 (td, J=7.6, 1.4 Hz, 1H), 7.28—7.23 (m, 1H), 6.05 (d, J=1.2 Hz, 1H), 5.60 (dd, J=3.2, 1.2 Hz, 1H), 5.23 (dd, J=10.0, 3.3 Hz, 1H), 5.13—5.06 (m, 3H), 5.00 (d, J=2.0 Hz, 1H), 3.98 (dq, J=9.7, 6.2 Hz, 1H), 3.79—3.68 (m, 2H), 2.20 (s, 3H), 2.14 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.00 (s, 3H), 1.54—1.48 (m, 1H), 1.45 (d, J=5.6 Hz, 3H), 1.23 (d, J=6.2 Hz, 3H), 0.95—0.85 (m, 4H) | 170.09, 170.05, 169.98, 169.91, 169.79, 163.25, 134.22, 132.23, 130.44, 129.98, 126.88, 125.49, 100.22, 99.36, 90.57, 78.30, 74.07, 73.21, 71.91, 70.86, 70.10, 69.02, 68.55, 67.41, 20.87, 20.82, 20.80, 20.68, 20.60, 18.06, 17.23, 8.99, 8.88, 0.66 |
| 8βa | 7.97 (ddd, J=7.9, 1.5, 0.5 Hz, 1H), 7.50 (dd, J=7.8, 1.2 Hz, 1H), 7.46 (td, J=7.6, 1.4 Hz, 1H), 7.36 (ddd, J=7.9, 7.2, 1.5 Hz, 1H), 6.25 (d, J=1.7 Hz, 1H), 5.48—5.41 (m, 2H), 5.14 (dd, J=9.9, 3.3 Hz, 1H), 5.11 (dd, J=3.2, 1.9 Hz, 1H), 5.08 (t, J=9.7 Hz, 1H), 5.02 (d, J=1.9 Hz, 1H), 4.17—4.10 (m, 1H), 3.96 (dq, J=9.4, 6.2 Hz, 1H), 3.81 (t, J=9.6 Hz, 1H), 2.15 (s, 3H), 2.14 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 1.98 (s, 3H), 1.56 (tt, J=8.2, 5.1 Hz, 1H), 1.40 (d, J=6.2 Hz, 3H), 1.22 (d, J=6.3 Hz, 3H), 0.95—0.84 (m, 4H) | 169.94, 169.92, 169.89, 169.79, 169.65, 163.80, 134.75, 132.34, 130.94, 130.01, 127.27, 125.20, 100.25, 99.45, 91.31, 78.32, 74.58, 71.46, 70.78, 69.91, 69.58, 69.13, 68.60, 67.42, 20.86, 20.80, 20.78, 20.66, 20.66, 18.20, 17.21, 9.07, 0.71 |
| 9a | 6.80 (d, J=8.2 Hz, 1H), 6.76 (d, J=1.8 Hz, 1H), 6.71 (dd, J=8.2, 1.7 Hz, 1H), 6.10—6.00 (m, 2H), 5.43—5.35 (m, 2H), 5.28—5.22(m, 2H), 4.58—4.52 (m, 4H), 4.34 (d, J=3.2 Hz, 2H), 4.30 (d, J=7.7 Hz, 1H), 4.04 (dt, J=15.5, 7.6 Hz, 1H), 4.00—3.81 (m, 3H), 3.75—3.66 (m, 1H), 3.52 (t, J=9.0 Hz, 1H), 3.48—3.44 (m, 1H), 3.43—3.35 (m, 2H), 2.92—2.79 (m, 2H), 2.06 (s, 3H) | 171.66, 148.45, 147.19, 133.56, 133.50, 131.07, 121.41, 117.51, 117.46, 115.33, 114.44, 102.71, 76.09, 73.79, 73.39, 71.03, 70.07, 70.05, 70.01, 63.55, 35.56, 20.91 |
| Compd. | 1H NMR (500 MHz), δ | 13C NMR (126 MHz), δ |
| 10a | 7.62 (d, J=15.8 Hz, 1H), 7.04 (d, J=2.0 Hz, 1H), 7.01 (dd, J=8.3, 2.0 Hz, 1H), 6.83 (d, J=8.3 Hz, 1H), 6.78—6.74 (m, 2H), 6.70 (dd, J=8.2, 1.9 Hz, 1H), 6.31 (d, J=15.9 Hz, 1H), 6.11—5.99 (m, 4H), 5.45—5.35 (m, 4H), 5.32—5.21 (m, 4H), 4.65—4.50 (m, 9H), 4.41 (dd, J=12.3, 2.0 Hz, 1H), 4.32 (d, J=7.7 Hz, 1H), 4.12—4.06 (m, 1H), 3.74—3.67 (m, 1H), 3.59 (t, J=9.0 Hz, 1H), 3.54—3.34 (m, 6H, 3OH), 2.92—2.81 (m, 2H) | 168.02, 150.88, 148.54, 148.47, 147.17, 145.97, 133.59, 133.53, 133.03, 132.84, 131.16, 127.24, 123.00, 121.37, 117.98, 117.90, 117.50, 117.45, 115.21, 114.89, 114.38, 113.32, 112.68, 102.91, 75.88, 74.27, 73.56, 71.07, 70.04, 69.94, 69.92, 69.89, 69.67, 63.28, 35.61 |
| 16aa | 6.82 (d, J=8.2 Hz, 1H), 6.76 (d, J=1.9 Hz, 1H), 6.73 (dd, J=8.2, 1.9 Hz, 1H), 6.12—6.02 (m, 2H), 5.42 (ddd, J=7.2, 3.1, 1.5 Hz, 1H), 5.38 (ddd, J=7.3, 3.1, 1.5 Hz, 1H), 5.34 (dd, J=3.4, 1.8 Hz, 1H), 5.31 (dd, J=9.9, 3.4 Hz, 1H), 5.28—5.23 (m, 2H), 5.16 (d, J=1.6 Hz, 1H), 5.06 (t, J=9.9 Hz, 1H), 4.61—4.55 (m, 4H), 4.48 (dd, J=12.1, 3.8 Hz, 1H), 4.29 (dd, J=12.1, 1.3 Hz, 1H), 4.23 (d, J=7.7 Hz, 1H), 4.21—4.17 (m, 1H), 4.14—4.08 (m, 1H), 3.67 (dt, J=9.5, 7.5 Hz, 1H), 3.59—3.54 (m, 1H), 3.49—3.40 (m, 3H), 2.88—2.81 (m, 2H), 2.14 (s, 3H), 2.14 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 1.21 (d, J=6.3 Hz, 3H) | 171.77, 170.15, 170.02, 169.95, 148.49, 147.19, 133.59, 133.54, 131.26, 121.31, 117.48, 117.46, 115.16, 114.45, 102.88, 98.41, 82.44, 74.03, 73.88, 71.03, 71.00, 70.09, 69.98, 69.71, 69.00, 68.65, 66.97, 63.17, 35.55, 20.94, 20.91, 20.80, 20.73, 17.36 |
| 16ca | 6.82 (d, J=8.2 Hz, 1H), 6.77 (d, J=1.9 Hz, 1H), 6.73 (dd, J=8.1, 2.0 Hz, 1H), 6.12—6.02 (m, 2H), 5.42 (dq, J=7.6, 1.6 Hz, 1H), 5.38 (dq, J=7.6, 1.6 Hz, 1H), 5.29—5.24 (m, 3H), 5.13—5.07 (m, 2H), 4.88 (d, J=1.9 Hz, 1H), 4.61—4.55 (m, 4H), 4.32 (d, J=1.8 Hz, 2H), 4.27 (d, J=7.8 Hz, 1H), 4.16—4.07 (m, 2H), 3.76 (s, 1H, OH), 3.69 (dt, J=9.5, 7.6 Hz, 1H), 3.61—3.53 (m, 3H), 3.39 (t, J=8.4 Hz, 1H), 2.91—2.82 (m, 2H), 2.41 (s, 1H, OH), 2.14 (s, 3H), 2.09 (s, 3H), 2.05 (s, 3H), 2.00 (s, 3H), 1.25 (d, J=6.3 Hz, 3H) | 170.72, 170.01, 169.82, 148.46, 147.16, 133.56, 133.50, 131.14, 121.28, 117.52, 117.49, 115.08, 114.32, 102.55, 98.91, 80.55, 74.99, 73.53, 72.29, 71.05, 70.64, 70.06, 70.02, 69.92, 68.35, 67.87, 62.46, 35.56, 20.85, 20.82, 20.75, 20.70, 17.34 |
| 16da | 6.82 (d, J=8.2 Hz, 1H), 6.77 (d, J=2.0 Hz, 1H), 6.74 (dd, J=8.2, 2.0 Hz, 1H), 6.12—6.01 (m, 2H), 5.42 (dq, J=9.1, 1.6 Hz, 1H), 5.39 (dq, J=9.1, 1.7 Hz, 1H), 5.33—5.23 (m, 5H), 5.18 (dd, J=10.1, 3.2 Hz, 1H), 5.12—5.06 (m, 3H), 4.95 (d, J=1.9 Hz, 1H), 4.61—4.55 (m, 4H), 4.45—4.35 (m, 3H), 4.30 (dd, J=12.0, 5.0 Hz, 1H), 4.27 (d, J=1.9 Hz, 1H), 4.16—4.08 (m, 1H), 4.04 (dt, J=9.2, 7.8 Hz, 1H), 3.72—3.61 (m, 3H), 3.46—3.35 (m, 2H, 1OH), 2.86 (t, J=8.1 Hz, 2H), 2.13 (s, 3H), 2.12 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.97 (s, 3H), 1.24 (d, J=6.4 Hz, 3H), 1.15 (d, J=6.3 Hz, 3H) | 171.16, 170.14, 170.11, 169.97, 169.85, 169.76, 169.70, 148.51, 147.23, 133.56, 133.47, 130.64, 121.32, 117.55, 117.50, 115.04, 114.39, 101.37, 99.17, 98.02, 88.90, 75.44, 73.34, 71.30, 71.15, 70.54, 70.04, 69.99, 69.64, 69.49, 68.99, 68.35, 67.99, 66.50, 63.39, 35.52, 20.92, 20.85, 20.84, 20.78, 20.75, 20.64, 17.34, 17.15 |
| 17a | 7.62 (d, J=15.9 Hz, 1H), 7.09—7.02 (m, 2H), 6.86 (d, J=8.3 Hz, 1H), 6.81 (d, J=8.2 Hz, 1H), 6.74 (d, J=1.5 Hz, 1H), 6.71 (d, J=8.1 Hz, 1H), 6.22 (d, J=15.9 Hz, 1H), 6.14—6.00 (m, 4H), 5.45—5.36 (m, 4H), 5.33—5.18 (m, 5H), 5.12 (dd, J=10.1, 3.3 Hz, 1H), 5.08 (dd, J=9.5, 8.0 Hz, 1H), 5.04—5.00 (m, 1H), 4.94 (t, J=10.0 Hz, 1H), 4.87 (d, J=1.4 Hz, 1H), 4.67—4.54 (m, 8H), 4.40 (d, J=8.0 Hz, 1H), 4.19 (dd, J=12.4, 4.8 Hz, 1H), 4.15 (dd, J=12.4, 2.9 Hz, 1H), 4.06 (dt, J=9.3, 6.3 Hz, 1H), 3.90 (t, J=9.4 Hz, 1H), 3.83 (dq, J=12.4, 6.2 Hz, 1H), 3.68—3.57 (m, 2H), 2.79 (t, J=6.7 Hz, 2H), 2.09 (s, 3H), 2.07 (s, 3H), 1.99 (s, 3H), 1.94 (s, 3H), 1.84 (s, 3H), 1.03 (d, J=6.2 Hz, 3H) | 170.73, 169.97, 169.43, 169.32, 165.47, 151.06, 148.57, 148.40, 147.06, 146.28, 133.67, 133.57, 132.96, 132.72, 131.62, 127.06, 122.94, 121.38, 118.04, 117.94, 117.43, 117.38, 115.32, 114.46, 114.41, 113.37, 112.83, 100.84, 98.87, 80.23, 72.36, 72.09, 70.78, 70.57, 70.14, 70.13, 70.00, 69.93, 69.68, 69.30, 68.52, 67.15, 62.42, 35.55, 20.86, 20.82, 20.67, 20.62, 20.58, 17.53 |
| Compd. | 1H NMR (500 MHz), δ | 13C NMR (126 MHz), δ |
| 18ab | 7.65 (d, J=15.9 Hz, 1H), 7.23 (d, J=1.9 Hz, 1H), 7.16 (dd, J=8.4, 1.8 Hz, 1H), 6.98 (d, J=8.4 Hz, 1H), 6.91 (d, J=1.9 Hz, 1H), 6.87 (d, J=8.2 Hz, 1H), 6.79 (dd, J=8.2, 1.9 Hz, 1H), 6.39 (d, J=15.9 Hz, 1H), 6.13—6.01 (m, 4H), 5.46—5.35 (m, 4H), 5.29—5.21 (m, 4H), 5.20 (d, J=1.6 Hz, 1H), 4.92 (t, J=9.5 Hz, 1H), 4.64—4.60 (m, 4H), 4.58—4.52 (m, 4H), 4.38 (d, J=7.9 Hz, 1H), 4.08 (dt, J=9.7, 7.2 Hz, 1H), 3.91 (dd, J=3.2, 1.8 Hz, 1H), 3.82 (t, J=9.2 Hz, 1H), 3.79—3.73 (m, 1H), 3.65—3.50 (m, 5H), 3.39 (dd, J=9.1, 8.0 Hz, 1H), 3.31—3.26 (m, 1H), 2.91—2.83 (m, 2H), 1.09 (d, J = 6.2 Hz, 3H) | 166.57, 150.96, 148.55, 148.45, 146.98, 145.91, 133.78, 133.72, 133.34, 133.11, 132.21, 127.50, 122.89, 121.41, 116.41, 116.33, 116.06, 116.02, 115.40, 114.80, 114.68, 113.43, 112.91, 102.79, 101.55, 80.06, 74.78, 74.62, 72.34, 70.92, 70.65, 70.50, 69.92, 69.69, 69.57, 69.32, 69.28, 68.97, 60.98, 35.24, 17.04 |
| 18bb | 7.59 (d, J=15.9 Hz, 1H), 7.16 (d, J=2.0 Hz, 1H), 7.03 (dd, J=8.4, 2.0 Hz, 1H), 6.92 (d, J=8.4 Hz, 1H), 6.84 (s, 1H), 6.72—6.69 (m, 2H), 6.38 (d, J=16.0 Hz, 1H), 6.12—5.97 (m, 4H), 5.46—5.33 (m, 4H), 5.29—5.17 (m, 5H), 4.61—4.55 (m, 4H), 4.51 (dd, J=11.8, 2.3 Hz, 1H), 4.50—4.41 (m, 4H), 4.37 (dd, J=12.0, 6.6 Hz, 1H), 4.34 (d, J=7.9 Hz, 1H), 4.05—3.92 (m, 3H), 3.75 (ddd, J=9.9, 8.1, 6.9 Hz, 1H), 3.70 (dd, J=9.5, 3.4 Hz, 1H), 3.60—3.55 (m, 1H), 3.53 (t, J=8.9 Hz, 1H), 3.43—3.37 (m, 2H), 3.35—3.31 (m, 1H), 2.88—2.77 (m, 2H), 1.25 (d, J=6.2 Hz, 3H) | 167.33, 150.79, 148.51, 148.29, 146.87, 145.16, 133.72, 133.67, 133.36, 133.11, 131.84, 127.40, 122.84, 121.31, 116.43, 116.31, 116.07, 116.05, 115.09, 114.90, 114.30, 113.21, 112.41, 103.03, 101.32, 82.54, 74.28, 73.92, 72.54, 70.92, 70.85, 70.80, 69.71, 69.50, 69.43, 69.21, 69.15, 68.61, 63.33, 35.44, 16.47 |
| 19aa | 7.68 (d, J=15.9 Hz, 1H), 7.11—7.06 (m, 2H), 6.87 (d, J=8.9 Hz, 1H), 6.80 (d, J=8.2 Hz, 1H), 6.75 (d, J=1.9 Hz, 1H), 6.72 (dd, J=8.1, 2.0 Hz, 1H), 6.36 (d, J=15.9 Hz, 1H), 6.13—6.01 (m, 4H), 5.45 (td, J=3.1, 1.5 Hz, 1H), 5.43—5.40 (m, 2H), 5.38 (ddd, J=6.3, 3.2, 1.6 Hz, 1H), 5.35—5.28 (m, 4H), 5.26 (ddd, J=4.3, 2.8, 1.4 Hz, 1H), 5.24 (ddd, J=4.3, 2.8, 1.3 Hz, 1H), 5.17 (d, J=1.6 Hz, 1H), 5.06 (t, J=9.9 Hz, 1H), 4.73 (dd, J=12.0, 2.6 Hz, 1H), 4.66—4.62 (m, 4H), 4.59—4.54 (m, 4H), 4.33 (d, J=12.0 Hz, 1H), 4.28—4.21 (m, 2H), 4.14 (dt, J = 9.6, 6.7 Hz, 1H), 3.67 (dt, J=9.5, 7.5 Hz, 1H), 3.62—3.57 (m, 1H), 3.50—3.44 (m, 3H), 3.42 (s, 1H, OH), 2.89—2.81 (m, 2H), 2.23 (d, J=2.2 Hz, 1H, OH), 2.14 (s, 3H), 2.04 (s, 3H), 1.98 (s, 3H), 1.21 (d, J=6.3 Hz, 3H) | 170.11, 170.02, 169.97, 168.04, 150.88, 148.55, 148.49, 147.18, 146.09, 133.58, 133.53, 133.01, 132.82, 131.24, 127.31, 123.05, 121.30, 117.99, 117.91, 117.48, 117.45, 115.12, 114.85, 114.42, 113.35, 112.70, 103.01, 98.34, 81.83, 74.49, 74.07, 71.07, 71.06, 70.07, 69.96, 69.94, 69.73, 69.70, 69.04, 68.48, 66.86, 62.93, 35.57, 20.91, 20.81, 20.73, 17.34 |
| 19ba | 7.64 (d, J=15.9 Hz, 1H), 7.07— 7.03 (m, 2H), 6.86 (d, J=8.1 Hz, 1H), 6.80 (d, J=8.2 Hz, 1H), 6.78 (d, J=2.0 Hz, 1H), 6.74 (dd, J=8.2, 2.0 Hz, 1H), 6.32 (d, J=15.9 Hz, 1H), 6.12—6.01 (m, 4H), 5.47—5.22 (m, 11H), 5.07 (t, J=9.7 Hz, 1H), 4.73 (dd, J=12.4, 3.6 Hz, 1H), 4.67—4.54 (m, 8H), 4.43 (d, J=7.7 Hz, 1H), 4.33—4.26 (m, 2H), 4.08 (dt, J=9.1, 7.8 Hz, 1H), 3.73—3.66 (m, 2H), 3.55 (dd, J=9.2, 7.7 Hz, 1H), 3.43 (ddd, J=9.7, 3.6, 2.1 Hz, 1H), 3.35 (t, J=9.4 Hz, 1H), 2.88 (t, J=8.1 Hz, 2H), 2.14 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.17 (d, J =6.3 Hz, 3H) | 170.43, 170.41, 170.00, 168.43, 150.92, 148.52, 148.48, 147.16, 146.31, 133.56, 133.48, 132.97, 132.80, 130.80, 127.14, 123.09, 121.30, 118.06, 117.97, 117.56, 117.50, 115.02, 114.64, 114.34, 113.26, 112.50, 101.87, 97.57, 76.89, 74.16, 71.32, 71.10, 70.03, 69.95, 69.88, 69.86, 69.74, 69.68, 69.27, 66.45, 62.92, 35.64, 21.00, 20.85, 20.82, 17.25 |
| 20aa | 7.67 (d, J=15.9 Hz, 1H), 7.10—7.05 (m, 2H), 6.86 (d, J=8.3 Hz, 1H), 6.79 (d, J=8.2 Hz, 1H), 6.75 (d, J=2.0 Hz, 1H), 6.72 (dd, J=8.2, 2.0 Hz, 1H), 6.36 (d, J=15.9 Hz, 1H), 6.13—6.00 (m, 4H), 5.47—5.35 (m, 5H), 5.32—5.22 (m, 5H), 5.20 (dd, J=10.0, 3.3 Hz, 1H), 5.12—5.09 (m, 2H), 5.07 (t, J=9.9 Hz, 1H), 4.95 (d, J=2.0 Hz, 1H), 4.69—4.61 (m, 5H), 4.59—4.54 (m, 4H), 4.38 (dd, J=12.3, 2.0 Hz, 1H), 4.24 (d, J=7.7 Hz, 1H), 4.21—4.15 (m, 1H), | 170.25, 170.01, 169.95, 169.90, 169.78, 167.89, 150.75, 148.49, 148.43, 147.09, 145.94, 133.58, 133.52, 133.01, 132.85, 131.26, 127.35, 123.04, 121.29, 118.02, 117.94, 117.54, 117.50, 114.99, 114.31, 113.26, 112.55, 103.01, 99.26, 98.41, 82.22, 79.05, 74.38, 73.97, 71.40, 71.09, 70.89, |
| Compd. | 1H NMR (500 MHz), δ | 13C NMR (126 MHz), δ |
| 20aa | 4.13 (dt, J=9.5, 6.8 Hz, 1H), 3.97 (dq, J=9.9, 6.3 Hz, 1H), 3.70—3.63 (m, 2H), 3.60—3.45 (m, 4H), 2.88—2.81 (m, 2H), 2.13 (s, 3H), 2.10 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.97 (s, 3H), 1.35 (d, J=6.2 Hz, 3H), 1.21 (d, J=6.2 Hz, 3H) | 70.07, 70.03, 69.93, 69.89, 69.87, 69.68,68.68, 68.48, 67.39, 67.27, 62.97, 35.58, 20.92, 20.89, 20.81, 20.76, 20.69, 18.05, 17.23 |
| 20ba | 7.65 (d, J=15.9 Hz, 1H), 7.08—7.03 (m, 2H), 6.87 (d, J=8.1 Hz, 1H), 6.82 (d, J=1.7 Hz, 1H), 6.81—6.76 (m, 2H), 6.32 (d, J=15.9 Hz, 1H), 6.13—6.00 (m, 4H), 5.47—5.22 (m, 10H), 5.19—5.15 (m, 2H), 5.13 (dd, J=3.3, 1.9 Hz, 1H), 5.04 (t, J=9.9 Hz, 1H), 4.96 (d, J=2.0 Hz, 1H), 4.74 (dd, J=12.4, 3.5 Hz, 1H), 4.67—4.52 (m, 8H), 4.44 (d, J=7.7 Hz, 1H), 4.29 (dd, J=12.4, 2.0 Hz, 1H), 4.27—4.22 (m, 1H), 4.12—4.05 (m, 1H), 3.96—3.88 (m, 1H), 3.76—3.63 (m, 3H), 3.52 (dd, J=9.2, 7.7 Hz, 1H), 3.48—3.42 (m, 2H), 3.36 (td, J=9.3, 3.5 Hz, 1H, OH), 3.04 (d, J=3.0 Hz, 1H, OH), 2.90 (t, J=8.0 Hz, 2H), 2.12 (s, 3H), 2.10 (s, 3H), 2.05 (s, 3H), 1.96 (s, 3H), 1.95 (s, 3H), 1.32 (d, J=6.2 Hz, 3H), 1.16 (d, J=6.3 Hz, 3H) | 170.54, 170.16, 169.98, 169.94, 169.66, 168.40, 150.89, 148.52, 148.47, 147.06, 146.26, 133.64, 133.55, 132.99, 132.81, 130.98, 127.18, 123.07, 121.49, 118.06, 117.97, 117.51, 117.42, 115.08, 114.70, 114.39, 113.28, 112.51, 101.97, 99.31, 97.69, 79.08, 77.38, 74.10, 71.67, 71.46, 70.78, 70.08, 70.03, 69.89, 69.87, 69.81, 69.69, 68.70, 67.31, 67.17, 62.92, 35.77, 20.95, 20.88, 20.79, 20.71, 20.65, 17.96, 17.23 |
| Acteoside(1)b | 7.59 (d, J=15.9 Hz, 1H), 7.05 (d, J=1.9 Hz, 1H), 6.96 (dd, J=8.2, 1.9 Hz, 1H), 6.78 (d, J=8.2 Hz, 1H), 6.70 (d, J=1.9 Hz, 1H), 6.67 (d, J=8.0 Hz, 1H), 6.57 (dd, J=8.0, 1.9 Hz, 1H), 6.27 (d, J=15.9 Hz, 1H), 5.19 (d, J=1.3 Hz, 1H), 4.92 (t, J=9.5 Hz, 1H), 4.38 (d, J=7.9 Hz, 1H), 4.08—4.02 (m, 1H), 3.92 (dd, J=3.0, 1.7 Hz, 1H), 3.82 (t, J=9.2 Hz, 1H), 3.76—3.69 (m, 1H), 3.65—3.50 (m, 5H), 3.39 (dd, J=8.9, 8.1 Hz, 1H), 3.30—3.26 (m, 1H), 2.85—2.76 (m, 2H), 1.09 (d, J=6.2 Hz, 3H) | 168.27, 149.79, 148.00, 146.84, 146.14, 144.68, 131.46, 127.66, 123.19, 121.24, 117.10, 116.50, 116.29, 115.21, 114.70, 104.22, 103.03, 81.63, 76.22, 76.06, 73.80, 72.36, 72.26, 72.06, 70.59, 70.42, 62.37, 36.58, 18.45 |
| Isoacteoside (4)b | 7.55 (d, J=15.9 Hz, 1H), 7.02 (d, J=1.6 Hz, 1H), 6.88 (dd, J=8.1, 1.7 Hz, 1H), 6.76 (d, J=8.2 Hz, 1H), 6.66 (d, J=1.6 Hz, 1H), 6.62 (d, J=8.0 Hz, 1H), 6.52 (dd, J=8.0, 1.7 Hz, 1H), 6.28 (d, J=15.9 Hz, 1H), 5.17 (s, 1H), 4.48 (dd, J=11.8, 1.8 Hz, 1H), 4.34 (dd, J=11.9, 5.6 Hz, 1H), 4.32 (d, J=7.9 Hz, 1H), 4.03—3.91 (m, 3H), 3.73—3.66 (m, 2H), 3.55—3.49 (m, 2H), 3.42—3.36 (m, 2H), 3.34—3.30 (m, 1H), 2.82—2.71 (m, 2H), 1.23 (d, J=6.2 Hz, 3H) | 169.1, 149.6, 147.2, 146.8, 146.1, 144.6, 131.3, 127.6, 123.1, 121.2, 117.0, 116.5, 116.3, 115.0, 114.7, 104.3, 102.7, 83.8, 75.7, 75.4, 73.9, 72.4, 72.3, 72.2, 70.3, 70.0, 64.6, 36.7, 17.8 |
| Ligupurpuroside J (5)b | 7.56 (d, J=15.9 Hz, 1H), 7.03 (d, J=1.9 Hz, 1H), 6.89 (dd, J=8.2, 1.9 Hz, 1H), 6.76 (d, J=8.2 Hz, 1H), 6.66 (d, J=1.9 Hz, 1H), 6.62 (d, J=8.0 Hz, 1H), 6.53 (dd, J=8.1, 1.9 Hz, 1H), 6.29 (d, J=15.9 Hz, 1H), 5.19 (s, 2H), 4.49 (dd, J=11.8, 1.8 Hz, 1H), 4.35—4.30 (m, 2H), 4.11 (dq, J=12.5, 6.2 Hz, 1H), 3.98—3.91 (m, 2H), 3.90—3.86 (m, 1H), 3.83 (dd, J=9.2, 3.3 Hz, 1H), 3.74—3.66 (m, 2H), 3.59 (dd, J=9.5, 3.3 Hz, 1H), 3.56—3.50 (m, 3H), 3.41—3.35 (m, 2H), 2.81—2.72 (m, 2H), 1.27 (d, J=6.2 Hz, 3H), 1.24 (d, J=6.2 Hz, 3H) | 169.1, 149.6, 147.2, 146.8, 146.1, 144.6, 131.3, 127.6, 123.1, 121.2, 117.0, 116.5, 116.3, 115.0, 114.8, 104.4, 103.2, 102.4, 83.4, 81.1, 75.8, 75.4, 73.8, 73.0, 72.9, 72.4, 72.3, 70.4, 70.3, 68.4, 64.6, 36.7, 18.5, 17.8 |
a. The solvent was CDCl3; b. the solvent was CD3OD.
3 结 论
采用条件温和的金(Ⅰ)催化糖苷化方法, 区域选择性地构建苯丙素苷中常见的α-(1→3)糖苷键, 缩短了合成路线并提高了合成效率. 以此为关键反应, 从已知化合物11出发, 分别以6步反应6%的收率, 4步反应24%的收率完成了苯丙素苷Acteoside(1)和Isoacteoside(4)的合成; 从已知二糖15出发, 以7步反应26%的收率完成了Ligupurpuroside J(5)的首次全合成. 这些化合物的合成验证了该苯丙素苷合成路线的通用性.
支持信息见
参考文献
DOI:10.1016/S0031-9422(00)94371-1 URL [本文引用: 1]
DOI:10.1016/j.foodchem.2011.11.037 URL [本文引用: 1]
Verbascoside is a natural antioxidant extracted from Pedicularis striata Pall (Jueyehesen). After being treated with 20 mumol/l verbascoside, the growth curve and mitotic index of human gastric adenocarcinoma MGc80-3 cells decreased remarkably, cell doubling time was delayed, the cellular growth inhibitory rate amounted to 53.2%, cell surface charge assayed by cell electrophoresis obviously changed, the electrophoresis rate dropped from 3.51 microns/s/v/cm to 2.74, i.e., the percent of retardation reached 28.4%. There was a 75% decrease of the tumorigenicity for the treated cells compared with the untreated cells inoculated subcutaneously in BALB/C nude mice. Scanning electron microscopy revealed that the microvilli on the surface of treated cells had been reduced obviously. It confirmed that verbascoside, similar to DMSO, could reverse MGc80-3 cells' malignant phenotypic characteristics and induced redifferentiation of MGc80-3 cells.
DOI:10.1021/np9703914
URL
PMID:9599250
[本文引用: 1]

Three new phenylpropanoid glycosides, named luteoside A (3), luteoside B (4), and luteoside C (5), were isolated together with the known compounds verbascoside (1) and isoverbascoside (2) from the roots of the medicinal plant Markhamia lutea. The structures of the new compounds were determined to be 1-O-(3, 4-dihydroxyphenyl)ethyl beta-D-apiofuranosyl(1-->2)-alpha-l-rhamnopyranosyl(1-->3)-4-O- caffeo yl-6-acetyl-beta-d-glucopyranoside, 1-O-(3,4-dihydroxyphenyl)ethyl beta-d-apiofuranosyl(1-->2)-alpha-l-rhamnopyranosyl(1-->3)-6-O- caffeo yl-beta-d-glucopyranoside, and 1-O-(3,4-dihydroxyphenyl)ethyl beta-D-apiofuranosyl(1-->2)-alpha-l-rhamnopyranosyl(1-->3)-6-O- ferulo yl-beta-d-glucopyranoside, respectively, on the basis of chemical and spectroscopic data. All five phenylpropanoid glycosides exhibited potent in vitro activity against respiratory syncytial virus.
DOI:10.1080/14786419.2014.955490
URL
PMID:25205114
[本文引用: 1]
Ethanol extracts of Stachys glutinosa L. (Lamiaceae) were investigated for antioxidative properties, as well as antiproliferative action on various cell lines. The antioxidant activities were investigated by ABTS (2,2'-azinobis-3-ethylbenzothiazoline-6-sulphonic acid) assay, DPPH (1,1-diphenyl-2-picrylhydrazyl) radical scavenging, beta-carotene/linoleic acid assay, scavenging of hydrogen peroxide (horseradish peroxidase test), superoxide anion scavenging, and hypochlorous acid scavenging (taurine test). The antioxidant activity was reported as IC50 and reveals antioxidant effects. Antiproliferative effects were measured in vitro on three cell lines: HepG2 (human hepatocarcinoma), MCF7 (breast human adenocarcinoma) and C2C12 (mouse myoblast) cell lines by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The ethanol extract induced variations in cell viability on all cell lines tested. At 200 mug/mL, the effects on cell viability were - 23%, - 27% and - 37%, respectively, for C2C12, MCF7 and HepG2.
DOI:10.1016/j.bmc.2010.01.047
URL
PMID:20159656
[本文引用: 1]
The methanolic extract from fresh stems of Cistanche tubulosa (Orobanchaceae) was found to show hepatoprotective effects against D-galactosamine (D-GalN)/lipopolysaccharide (LPS)-induced liver injury in mice. From the extract, three new phenylethanoid oligoglycosides, kankanosides H(1) (1), H(2) (2), and I (3), were isolated together with 16 phenylethanoid glycosides (4-19) and two acylated oligosugars (20, 21). The structures of 1-3 were determined on the basis of spectroscopic properties as well as of chemical evidence. Among the isolates, echinacoside (4, IC(50)=10.2 microM), acteoside (5, 4.6 microM), isoacteoside (6, 5.3 microM), 2'-acetylacteoside (8, 4.8 microM), and tubuloside A (10, 8.6 microM) inhibited D-GalN-induced death of hepatocytes. These five isolates, 4 (31.1 microM), 5 (17.8 microM), 6 (22.7 microM), 8 (25.7 microM), and 10 (23.2 microM), and cistantubuloside B(1) (11, 21.4 microM) also reduced TNF-alpha-induced cytotoxicity in L929 cells. Moreover, principal constituents (4-6) exhibited in vivo hepatoprotective effects at doses of 25-100mg/kg, po.
DOI:10.1016/S0008-6215(98)00092-5 URL [本文引用: 1]
DOI:10.1021/jo9906983 URL [本文引用: 1]
DOI:10.1007/BF00766648
URL
[本文引用: 2]
The total chemical synthesis of isoacteoside (1), 2-(3′,4′-dihydroxyphenyl)ethyl 6-O-caffeoyl-3-O-(α-l-rhamnopyranosyl)-β-d-glucopyranoside, is described. An acteoside acetate with benzyl groups at the catechols (3: 2-(3′,4′-dibenzyloxyphenyl)ethyl 2,6-di-O-acetyl-4-O-[3′,4′-bis(O-benzyl)caffeoyl]-3-O-(α-l-rhamnopyranosyl)-β-d-glucopyranoside) was treated with a solution of methy-lamine in methanol (MeNH2 in MeOH) to perform both deacetylation and caffeoyl migration, affording an isoacteoside derivative with benzyl groups at the catechols4b: 2-(3′,4′-dibenzyloxyphenyl)ethyl 6-O-[3′,4′-bis(O-benzyl) caffeoyl] -3-O-(α-l-rhamnopyranosyl)-β-d-glucopyranoside —in 34% yield. Debenzylation of4b was successfully accomplished by catalytic transfer hydrogenation using 1,4-cyclohexadiene to give the target compound isoacteoside (1) in 54% yield.1H and13C nuclear magnetic resonance spectral data of the synthesized isoacteoside (1) were identical with those of the natural isoacteoside isolated fromPaulownia tomentosa (Thumb.) Steud.]]>
DOI:10.1016/j.tetlet.2008.04.017 URL [本文引用: 3]
DOI:10.1021/ja4064316
URL
PMID:24252170
[本文引用: 1]
Anomerization, which involves cleavage and formation of the anomeric C-O bond, is of fundamental importance in the carbohydrate chemistry. Herein, the unexpected gold(I)-catalyzed anomerization of glycosyl ortho-alkynylbenzoates has been studied in detail. Especially, crossover experiments in the presence of an exogenous isochromen-4-yl gold(I) complex confirm that the anomerization proceeds via the exocleavage mechanism, involving (surprisingly) the addition of the isochromen-4-yl gold(I) complex onto a sugar oxocarbenium (or dioxolenium) and an elimination of LAu(+) from the vinyl gold(I) complex. The inhibitory effect of the exogenous isochromen-4-yl gold(I) complex when in stoichiometric amount on the anomerization has guided us to disclose an isochromen-4-yl gem-gold(I) complex, which is inactive in catalysis but in equilibrium with the monogold(I) complex and the LAu(+) catalyst. The proposed key intermediate in the anomerization, a transient glycosyloxypyrylium species, is successfully trapped via a cycloaddition reaction with n-butyl vinyl ether as a dienophile. SN2-like substitution of the initially formed glycosyloxypyrylium intermediate has then been achieved to a large extent via charging with acceptors in an excess amount to lead to the corresponding glycosides in a stereoselective manner.
DOI:10.1039/c8cs00209f
URL
PMID:29993057
[本文引用: 1]
The methodological developments in gold(i)- and gold(iii)-catalyzed glycosylation reactions are fully surveyed, which exploit the special alkynophilicity or the Lewis acidity of the gold cationic complexes. The application of the new methods in the total synthesis of naturally occurring glycoconjugates and glycans is comprehensively reviewed, with a focus on glycosylation of various complex aglycones.
DOI:10.1021/acs.accounts.7b00573
URL
PMID:29297680
[本文引用: 1]
Naturally occurring glycans and glycoconjugates have extremely diverse structures and biological functions. Syntheses of these molecules and their artificial mimics, which have attracted the interest of those developing new therapeutic agents, rely on glycosylation methodologies to construct the various glycosidic linkages. In this regard, a wide array of glycosylation methods have been developed, and they mainly involve the substitution of a leaving group on the anomeric carbon of a glycosyl donor with an acceptor (a nucleophile) under the action of a particular promoter (usually a stoichiometric electrophile). However, glycosylations involving inherently unstable or unreactive donors/acceptors are still problematic. In those systems, reactions involving nucleophilic, electrophilic, or acidic species present on the leaving group and the promoter could become competitive and detrimental to the glycosylation. To address this problem, we applied the recently developed chemistry of alkynophilic gold(I) catalysts to the development of new glycosylation reactions that would avoid the use of the conventional leaving groups and promoters. Gratifyingly, glycosyl o-alkynylbenzoates (namely, glycosyl o-hexynyl- and o-cyclopropylethynylbenzoates) turned out to be privileged donors under gold(I) catalysis with Ph3PAuNTf2 and Ph3PAuOTf. The merits of this new glycosylation protocol include the following: (1) the donors are easily prepared and are generally shelf-stable; (2) the promotion is catalytic; (3) the substrate scope is extremely wide; (4) relatively few side reactions are observed; (5) the glycosylation conditions are orthogonal to those of conventional methods; and (6) the method is operationally simple. Indeed, this method has been successfully applied in the synthesis of a wide variety of complex glycans and glycoconjugates, including complex glycosides of epoxides, nucleobases, flavonoids, lignans, steroids, triterpenes, and peptides. The direct glycosylation of some sensitive aglycones, such as dammarane C20-ol and sugar oximes, and the glycosylation-initiated polymerization of tetrahydrofuran were achieved for the first time. The gold(I) catalytic cycle of the present glycosylation protocol has been fully elucidated. In particular, key intermediates, such as the 1-glycosyloxyisochromenylium-4-gold(I) and isochromen-4-ylgold(I) complexes, have been unambiguously characterized. Exploiting the former glycosyloxypyrylium intermediate, SN2-type glycosylations were realized in specific cases, such as beta-mannosylation/rhamnosylation. The protodeauration of the latter vinylgold(I) intermediate has been reported to be critically important for the gold(I) catalytic cycle. Thus, the addition of a strong acid as a cocatalyst can dramatically reduce the required loading of the gold(I) catalyst (down to 0.001 equiv). C-Glycosylation with silyl nucleophiles can proceed catalytically when moisture, which is sequestered by molecular sieves, can serve as the H(+) donor for the required protodeauration step. Indeed, the unique mechanism explains the merits and broad applicability of the present glycosylation method and provides a foundation for future developments in glycosylation methodologies that mainly involve improving the diastereoselectivity and catalytic efficiency of glycosylations.
DOI:10.6023/A17120544
URL
[本文引用: 1]
1) and Notoginsenoside R1 (2) are two representative dammarane type protopanaxatriol-6,20-O-bisglycosides occurring widely founded in Panaxginseng. Ginsenoside Re (1) showed potent antioxidative, anti-inflammatory and antihyperlipemia activities, and Notoginsenoside R1 (2) showed potent antioxidative and antiinflammatory activities, so it would be helpful to synthesize these homogeneous natural products in appreciable amounts by accelerating their structure-activity relationship study. As a persistent effort on the chemical syntheses of the diverse ginsenosides in our group, we report herein the efficient syntheses of these two complex natural products. Thus, based on the reactivity sequence of the four hydroxyl groups (i.e., 12-OH > 3-OH > 6-OH >> 20-OH) of the protopanaxatriol aglycon, an orthogonal and efficient protecting group strategy was applied to distinguish these hydroxyl groups. The subsequent installation of the 6,20-O-bisglycosides are challenging, given the poor reactivity of the secondary 6-OH and tertiary 20-OH, moreover, with the latter being labile toward acidic conditions. Taking advantage of the neutral conditions of the Au(I)-catalyzed glycosylation reaction (0.3 equiv. Ph3PAuNTf2, 4 Å MS, CH2Cl2, r.t.), the glycosylation of the acid-labile 20-hydroxyl group was achieved effectively in a high 84% yield firstly. To be convergent for the syntheses of these two ginsenosides, the 6-O-disaccharide residues were installed in a stepwise manner. For the glycosylation of the 6-OH of protopanaxatriol, a higher loading of the catalyst Ph3PAuNTf2 (0.5 equiv.) was employed in order to increase the glycosylation yield while reduce the orthoester formation, thus, the desired 6β-O-glucosides were prepared in satisfactory yields (77%~83%). Both terminal α-L-Rha/β-D-Xyl moieties at the 2' position of 6-O-glc were installed efficiently under 0.2 equiv. Ph3PAuNTf2 catalyzing condition (ClCH2CH2Cl, 5 Å MS, 40℃) with 86% and 81% yields, respectively. After global deprotection, Ginsenoside Re (1) and Notoginsenoside R1 (2) were synthesized with the longest 13 linear steps in 5.1% and 4.5% overall yields, respectively.]]>
DOI:10.6023/A17120544
URL
[本文引用: 1]
O-Glc'糖基2位羟基连续三次糖苷化,以最长线性13步分别以5.1%和4.5%的收率合成了天然双糖基达玛烷皂苷Ginsenoside Re(1)和Notoginsenoside R1(2).]]>
DOI:10.6023/A19060233
URL
[本文引用: 1]
O-β-D-glucuronide (1) and scutellarin (scutellarein-7-O-β-D-glucuronide, 2) are two major flavone glucuronide components occurring in breviscapines, which are prepared from the traditional Chinese herb Erigeron breviscapus. These two flavone glycosides show potent anti-oxidative, anti-inflammatory and neuroprotective activities in various evaluations. Synthesis of these natural glycosides in an efficiently manner would facilitate studies on their structure activity relationships. As a persistent effort on the chemical syntheses of the diverse glycoconjugates from traditional Chinese herbs in our group, we report herein the synthesis of these two representative flavone O-glucuronides. It is known that the solubility of flavone compounds is rather low and this property would greatly hinder their glycosylation reactions. In order to increase the solubility of the flavone derivatives in the glycosylation solvents, hexanoyl and benzyl groups were selected as the permanent protecting groups for the hydroxyl groups of apigenin (7) and scutellarein (8). The construction of the phenolic O-glucuronide is known to be a difficult task, especially the glycosylation of the poorly nucleophilic 7-hydroxyl group which locates at the para-position of the flavone carbonyl group. We achieved the glycosylation of the flavone 7-OH with 2,3,4-tri-O-benzoyl-6-O-TBDPS-glucopyranosyl ortho-alkynylbenzoate (9) under the catalysis of Ph3PAuNTf2 (0.2 equiv., 4 ? MS, CH2Cl2, r.t., 5 h) in excellent yields. After that, the 6-O-TBDPS groups were removed, and the requisite glucuronides were then elaborated by oxidation of the resulting 6-OH under the conditions of DAIB/TEMPO (CH2Cl2/H2O, V∶V=2∶1, r.t.) in good yields. After global deprotection, the desired products apigenin-7-O-β-D-glucuronide (1) and scutellarin (2) were obtained in overall yields of 36% (5 steps) and 7% (9 steps), respectively, from the starting flavone aglycones. Following the same strategy, four naturally occurring flavone-7-O-glycosides, namely apigetrin (3), plantaginin (4), apigenin 7-O-β-D-xylopyranoside (5) and apigenin 7-O-α-L-rhamnopyranoside (6), were smoothly synthesized in 4~7 steps with the overall yields of 61%, 13%, 58% and 61%, respectively.]]>
DOI:10.6023/A19060233
URL
[本文引用: 1]
O-β-D-glucuronide, 1)和乙素(scutellarin, scutellarein-7-O-β-D-glucuronide, 2)是灯盏花素(breviscapine)中的两种主要黄酮苷成分, 具有抗氧化、抗肿瘤和治疗老年痴呆等生理活性; 大波斯菊苷(apigetrin, 3)、车前子苷(plantaginin, 4)、apigenin 7-O-β-D-xylopyranoside (5)、apigenin 7-O-α-L-rhamnopyranoside (6)等黄酮-7-O-糖苷也具有相似的结构和生理活性. 本工作针对黄酮苷元(芹菜素7和野黄芩素8)溶解度差、7位羟基酸性强而亲核性较弱以及糖醛酸糖基化给体反应活性较弱的问题, 综合利用使苷元有效增溶的保护基策略、金(I)催化的糖苷化方法和后期糖醛酸氧化策略, 高效构建了黄酮-7-O-葡萄糖醛酸结构, 并经统一的保护基脱除完成了灯盏花甲素(1) (36%)和乙素(2) (7%)的合成. 采用相似的策略, 从苷元出发分别以4~7步完成了天然黄酮-7-O-糖苷3~6的合成.]]>
DOI:10.1002/cjoc.v37.8 URL [本文引用: 1]
DOI:10.1002/anie.v58.31 URL [本文引用: 1]
DOI:10.1002/anie.v57.52 URL [本文引用: 2]
DOI:10.1039/C4RA03954H URL [本文引用: 2]
DOI:10.1021/jacs.9b00210
URL
PMID:30864790
[本文引用: 4]
A highly effective and versatile glycosylation method is developed, which uses 3,5-dimethyl-4-(2'-phenylethynylphenyl)phenyl (EPP) glycosides as donors and NIS/TMSOTf as promoter and proceeds via an unprecedented dearomative activation mechanism.
DOI:10.1039/c2ob25124h
URL
PMID:22678124
[本文引用: 2]
We report on a diastereoselective synthesis of six derivatives of caffeoyl- and feruloyl-muco-quinic acids. All the muco-quinic acid derivatives were obtained in excellent yield in five steps starting from quinic acid, caffeic acid and ferulic acid. Allyl ether protection of trans-hydroxy cinnamic acids was here introduced to chlorogenic acids synthesis. We show that muco-quinic acid derivatives, which are formally diastereoisomers of chlorogenic acids, can be readily distinguished by their tandem mass spectra.
DOI:10.1016/j.tetlet.2010.10.135
URL
[本文引用: 1]
AbstractTreatment of saccharidic polyols in ethyl acetate with catalytic sulfuric acid leads to the corresponding primary monoacetate derivatives in good yields. The transesterification was realized by simple stirring without rigorous exclusion of moisture or oxygen. Our protocol is applicable to the regioselective monoacetylation of amino sugars having different substituents at the 2-positions.Graphical abstract]]>

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}