天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1 实验部分
1.1 试剂与仪器
锡粉和硝酸镧[La(NO3 )3 ·6H2 O]均为分析纯, 购于天津市风船化学试剂科技有限公司; 硝酸钴[Co(NO3 )2 ·6H2 O]和硝酸铈[Ce(NO3 )3 ·6H2 O] 均为分析纯, 购于国药集团化学试剂有限公司; 浓硝酸(HNO3 , 质量分数为65%), 天津市北联精细化学品开发有限公司.
D8ADVANCE型X射线粉末衍射(XRD, 德国Bruker公司); ASAP2020M比表面孔径测定仪(BET, 美国Micromeritics公司); TecnaiG2F20S-Twin200KV型场发射透射电镜(TEM, 美国FEI公司); Multimode型原子力显微镜(AFM, 美国Veeco公司); CBT-1型程序升温还原(H2 -TPR, 美国Quantachrome Instrument公司); Topologic Systems MFD-500A型穆斯堡尔谱测试仪(日本Topologic Systems公司); Thermo EACALAB 250型X光电子能谱仪(XPS, 美国thermo scientific公司).
1.2 催化剂的制备
采用溶胶凝胶法制备单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 3 )3 ·6H2 O和Co(NO3 )2 ·6H2 O溶液, 继续回流4 h后, 将溶液转移至烧杯中于70 ℃恒温搅拌7 h, 形成凝胶; 将前驱体在100 ℃下干燥24 h, 于500 ℃马弗炉中分解3 h, 再于900 ℃马弗炉中焙烧3 h, 得到未负载催化剂La2 Sn1.7 Co0.3 O7- δ
采用溶胶凝胶-浸渍法制备烧绿石型逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3 )3 ·6H2 O和Co(NO3 )2 ·6H2 O溶液, 以及适量的柠檬酸溶液, 继续于90 ℃回流反应4 h; 将溶液转移至烧杯, 同时加入制备好的CeO2 粉体(CeO2 质量分数为30%), 于70 ℃恒温下搅拌7 h, 形成凝胶; 将前驱体在100 ℃下干燥24 h, 于500 ℃马弗炉中焙烧3 h, 再于900 ℃马弗炉中焙烧3 h, 得到烧绿石型逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ
机械混合对比样品CeO2 -La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 按照质量比7∶3进行简单机械混合, 制备出机械混合对比样品CeO2 -La2 Sn1.7 Co0.3 O7- δ
采用溶胶凝胶法制备稀土氧化物CeO2 . 称取计量比的Ce(NO3 )3 ·6H2 O和柠檬酸, 加适量去离子水溶解后置于烧杯中, 于70 ℃下搅拌7 h, 100 ℃下烘干, 于500 ℃马弗炉中焙烧2 h, 再于900 ℃马弗炉中焙烧3 h, 制成CeO2 样品(记为样品d).
1.3 催化剂的活性测试
采用常压固定床石英反应器(直径10 mm)进行催化剂活性测试. 催化剂颗粒为40~60 目, 用量为200 mg. 反应混合气CH4 /O2 /N2 的体积比为2∶18∶80. 反应时气体的总流速为40 mL/min, 空速为48000 h-1 (质量流量计控制). 用程序升温控制仪控制反应温度, 用FQ-W型CH4 红外分析仪分析不同温度下CH4 的转化率, 以表征催化剂的催化燃烧活性.
2 结果与讨论
2.1 XRD分析
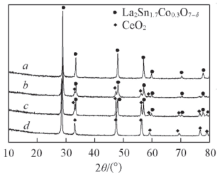
图1 为催化剂经900 ℃煅烧后的XRD谱图. 可见利用溶胶凝胶法制备的纯稀土氧化物(样品d)为单相CeO2 物相(PDF No.34-0394).
Fig.1 XRD patterns of the catalystsa. La2 Sn1.7 Co0.3 O7- δ b. CeO2 /La2 Sn1.7 Co0.3 O7- δ c. CeO2 -La2 Sn1.7 Co0.3 O7- δ d. CeO2 .
样品a在2θ 为29.0°, 36.6°和57.1°的衍射峰对应烧绿石的晶面(222), (331)和(622)的特征衍射峰, 且未检测到其它物相特征峰, 表明物相为单一的La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的(111), (200), (220)和(311)晶面所对应的特征衍射峰, 说明此样品由La2 Sn1.7 Co0.3 O7- δ 2 2种物相组成. 样品c中同样也出现了La2 Sn1.7 Co0.3 O7- δ 2 2种物相, 但值得注意的是, 机械混合样品c的CeO2 衍射峰强度比逆负载催化剂b的更强, 说明逆负载催化剂b中CeO2 在La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的负载对La2 Sn1.7 Co0.3 O7- δ 表1 . 由表1 可见, 逆负载催化剂b与单一烧绿石型催化剂a相比, La2 Sn1.7 Co0.3 O7- δ 2 的负载能够有效阻止La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的影响弱于烧绿石逆负载型催化剂. 通过公式[11 ] : (2ω )2 cos2 θ =4/π 2 (λ /Dhkl )2 +32< ε 2 h kl 2 θ , 计算相关催化剂中烧绿石晶格的扭曲程度, 其中Dhkl 是晶面间距; λ 为X射线波长(λ =0.15406 nm); θ 为晶面(hkl )的衍射角; 2ω 为校正的衍射峰半高宽(FWHM); < ε 2 h kl 1/2 为晶面扭曲度, 计算结果列于表1 . 逆负载催化剂b中的烧绿石晶体结构发生了较大程度的扭曲, 表明CeO2 对La2 Sn1.7 Co0.3 O7- δ [12 ] .
2.2 催化活性的测试
图2 (A)为催化剂的甲烷燃烧催化活性测试结果. T 90 表示通过催化剂床层的甲烷气体转化90%时的温度, 可以看出, 烧绿石逆负载型催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 (样品d). 单一烧绿石型催化剂(样品a)的T 90 为642 ℃, 而烧绿石逆负载型催化剂(样品b)的T 90 仅为564 ℃, 比单一烧绿石型催化剂降低了78 ℃. 同时, 与机械混合催化剂比较发现, 机械混合催化剂T 90 高达699 ℃, 催化活性远不如CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的逆负载作用.
Fig.2 Catalytic performance of catalysts(A), T 90 of catalysts with different calcination temperature of 900, 1000, 1100 and 1200 ℃(B), the cycling runs tests(C) and XRD patterns before(a ) and after(b ) methane oxidation reaction of inverse CeO2 /La2 Sn1.7 Co0.3 O7- δ a. La2 Sn1.7 Co0.3 O7- δ b. CeO2 /La2 Sn1.7 Co0.3 O7- δ c. CeO2 -La2 Sn1.7 Co0.3 O7- δ d . CeO2 .
将催化剂分别在900, 1000, 1100和1200 ℃下焙烧, 其高温热稳定性结果如图2 (B)所示. 烧绿石逆负载型催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ T 90 变化范围很小, 其在1200 ℃下焙烧的T 90 仅比900 ℃焙烧时升高了38 ℃, 而单一烧绿石和机械混合催化剂的T 90 变化均较大, 温度变化斜率依次递增, 在900~1200 ℃的范围内, T 90 分别升高89和138 ℃, 可见单一烧绿石和机械混合催化剂的高温热稳定性能均不如逆负载型催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 图2 (C)给出催化剂连续5次循环使用的活性测试结果. 可以看出, 单一烧绿石催化剂在5次连续测试后, 甲烷转化温度升高21 ℃, 较第一次反应时的催化活性明显降低, 而逆负载型催化剂使用5次后的T 90 却只升高了6 ℃, 图2 (D)给出烧绿石逆负载催化剂在5次连续反应测试前后的XRD谱图. 可见测试前后催化剂结构没有发生改变, 仍保留了La2 Sn1.7 Co0.3 O7- δ 2 的完整结构, 且晶体出峰位置和强度大小均没有变化. 可见, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ
2.3 比表面积(BET)分析
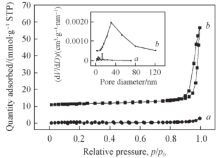
单一烧绿石催化剂和烧绿石型逆负载催化剂的吸附-脱附曲线及孔径分布如图3 所示. 2个样品吸附-脱附等温线均呈现高压段上扬的趋势, 烧绿石型逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7 吸附氮气的能力较La2 Sn1.7 Co0.3 O7- δ 2 的添加有效改善了催化剂吸附氮气的能力, 扩大了催化剂的孔分布范围, 同时, 增大了催化剂的孔容、 孔径和比表面积(表2 ), 从而有利于提高催化剂的催化性能. 此外, 表2 中还给出了机械混合催化剂CeO2 -La2 Sn1.7 Co0.3 O7- δ 2 的比表面积、 孔容和孔径大小. 单一烧绿石La2 Sn1.7 Co0.3 O7- δ 2 /g, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7 的比表面积为6.1 m2 /g, 而机械混合法制备的CeO2 -La2 Sn1.7 Co0.3 O7- δ 2 /g, 纯CeO2 的比表面积为7.5 m2 /g, 显然, 机械混合法制备的CeO2 -La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 的比表面数据反映出机械混合法制备的CeO2 -La2 Sn1.7 Co0.3 O7 催化剂中, CeO2 没有很好地发挥出增大催化剂比表面的作用.
Fig.3 N2 adsorption-desorption isotherms of La2 Sn1.7 Co0.3 O7- δ a ) and CeO2 /La2 Sn1.7 Co0.3 O7- δ b )
2.4 透射电子显微镜(TEM)和原子力显微镜(AFM)分析
图4 为催化剂样品的透射电子显微镜(TEM)、 高分辨透射电子显微镜(HRTEM)和原子力显微镜(AFM)照片. 图4 (A)~(C)分别是单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 图4 (A), (C)可见, 催化剂大部分颗粒聚集在一起, 表面发生严重烧结, 颗粒分布在40~70 nm之间, 由图4 (B)可观察到样品的几个晶面均为具有烧绿石复合氧化物La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 图4 (D)~(F)为CeO2 /La2 Sn1.7 Co0.3 O7- δ 图4 (D)中呈现出大小不同且相互交错的颗粒形貌, 由图4 (E)的HRTEM照片可确定, 晶面间距为0.31 nm的晶面, 对应烧绿石型晶体La2 Sn1.7 Co0.3 O7- δ 2 的(220)晶面, 故可确定具有较小粒径的颗粒为CeO2 粒子, 具有较大粒径的颗粒为La2 Sn1.7 Co0.3 O7- δ 2 粒子负载到较大的La2 Sn1.7 Co0.3 O7- δ 2 /CuO催化剂的颗粒形貌相似[13 ] . 根据TEM照片可测量出CeO2 颗粒尺寸在5~11 nm之间, 烧绿石样品粒径在20~50 nm之间, 较单一La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载烧绿石La2 Sn1.7 Co0.3 O7- δ
Fig.4 TEM(A,D), HRTEM(B, E) and AFM(C, F) analyses of La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ
2.5 H2 程序升温还原(H2 -TPR)分析
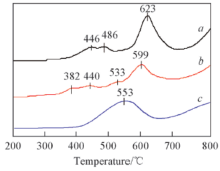
图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别.
Fig.5 TPR spectra of La2 Sn1.7 Co0.3 O7- δ a ), CeO2 /La2 Sn1.7 Co0.3 O7- δ b ) and CeO2 (c )
2.6 穆斯堡尔谱分析
图6 给出了单一烧绿石催化剂La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ
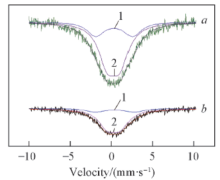
Fig.6 Mössbauer spectra of La2 Sn1.7 Co0.3 O7- δ a ) and CeO2 /La2 Sn1.7 Co0.3 O7- δ b )
由图6 可知, 2个样品中均有2种亚谱, 化学位移均在零速度附近, 且均为四极分裂双峰, 可判定亚谱中的锡均为Sn4+ , 故2种催化剂中Sn的价态主要以+4价存在. 另外, 根据文献[20 ]报道的SnO2 穆斯堡尔谱分析, 将催化剂中的亚谱1归属为界面相的Sn4+ , 亚谱2归属为结晶相的Sn4+ . 表3 列出了各样品的穆斯堡尔参数, 单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 4+ 含量降低, 结晶相Sn4+ 含量升高, 这可能正是甲烷燃烧催化活性提高的原因之一. 此外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 相互作用, 致使Sn核处的s 电子密度变小, 从而导致产生的IS值变大; 另外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的影响, 致使烧绿石晶体结构发生一定程度的畸变[21 ] , 促进甲烷燃烧催化活性的提高.
2.7 X射线光电子能谱及反应机理分析
图7 为催化剂的X射线光电子能谱. 用C1 s 图7 (A)给出单一烧绿石催化剂和逆负载催化剂的O1 s 1 s [22 ~24 ] , 通常将528~530 eV低结合能处的峰归属为晶格氧, 531~533 eV高结合能处的峰归属为吸附氧. 单一La2 Sn1.7 Co0.3 O7- δ [25 ] , 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 使La2 Sn1.7 Co0.3 O7- δ 2 与La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 没有明显影响到催化剂的吸附氧结合能变化, 但却使催化剂晶格氧的结合能发生了较明显的偏移. 表4 列出了催化剂样品O1 s 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载到La2 Sn1.7 Co0.3 O7- δ
Fig.7 XPS spectra of O1 s 3 d 3 d 2 Sn1.7 Co0.3 O7- δ a ) and CeO2 /La2 Sn1.7 Co0.3 O7- δ b )
图7 (B)为催化剂中Sn3 d 2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 4+ 结合能峰[26 ,27 ] , 故可确定样品中Sn价态主要为+4价, 与穆斯堡尔谱测试结果相符. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 与烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 改变了Sn周围的电子云密度, 使Sn周边的化学环境发生变化; 而且CeO2 的负载降低了催化剂中Sn3 d [28 ] , 从而使逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 表4 可知, La2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 2 /La2 Sn1.7 Co0.3 O7- δ 3 d 2 的负载对烧绿石结构中S n 3 d 3 2 n 3 d 5 2 3 d 图7 (C), L a 3 d 3 2 a 3 d 5 2 3+ 的结合能没有受到CeO2 的影响. 由于添加的钴含量过少, 在XPS测试中钴的结合能较微弱, 因此, 不能明确确定CeO2 的影响.
在没有添加催化剂的情况下, 甲烷燃烧反应的机理为自由基反应机理, 自由基剧烈运动, 会导致甲烷反应温度的急剧上升, 高达2000 ℃以上. 加入催化剂后, 甲烷燃烧反应机理变得十分复杂, 此时, 不但会发生自由基反应, 而且还会发生表面氧化反应. 对于耐高温性较好的烧绿石型复合氧化物催化剂, 催化剂表面吸附氧和晶格氧是影响催化剂活性的主要因素, 通常将甲烷催化燃烧反应分为“面上反应(suprafacial)”和“面内反应(intrafacial)”两种反应机制[29 ] , 在温度较低时一般是吸附氧起主要作用, 属于“面上反应”机制, 在温度较高时一般是晶格氧起主要作用, 属于“面内反应”机制. 用耐高温性较好的烧绿石复合氧化物La2 Sn1.7 Co0.3 O7- δ
式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为
O 2 - ( lat ) + 2 C e 4 + 1 2 O 2 ( Ce O 2 ) + Vo + 2 C e 3 + ( 3 ) 2 O 2 ( Ce O 2 ) + C H 4 C O 2 + 2 H 2 O 4 2 C e 3 + + Vo + 1 2 O 2 amb 2 C e 4 + + O lat 2 - 5
式中, O 2 ( Ce O 2 ) 2 自身释放出来的O2 . 由于CeO2 具有较强的储放氧功能[35 ] , 将CeO2 应用于甲烷催化燃烧反应, CeO2 中的部分氧可以释放出来用于氧化甲烷, 而自身变成Ce2 O3 , Ce2 O3 在反应气提供的O2 过剩的条件下又恢复成CeO2 . 因此, 我们认为逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ
综上所述, 通过溶胶凝胶-浸渍法制备了烧绿石逆负载型催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 可实现甲烷燃烧催化剂的优化.
参考文献
文献选项
[1]
Hayes R. E. Chem. Eng. Sci. , 2004 , 59 (19 ), 4073 —4080
[本文引用: 1]
[2]
Pan X. Q. Zhang Y. B. Zhang B. Miao Z. Z. Wu T. X. Yang X. G. Chem. Res. Chinese Universities , 2013 , 29 (5 ), 952 —955
[本文引用: 1]
[3]
Lee J. H. Trimm D. L. Fuel Process. Technol. , 1995 , 42 (2/3 ), 339 —359
[本文引用: 1]
[4]
Müller C. A. Maciejewski M. Koeppel R. A. Baiker A. Catal. Today , 1999 , 47 (1—4 ), 245 —252
[5]
Spinicci R. Tofanari A. Appl. Catal. A: Gen. , 2002 , 227 (1/2 ), 159 —169
[6]
Eguchi K. Arai H. Catal. Today , 1996 , 29 (1 ), 379 —386
[7]
Prasad R. Kennedy L. A. Ruckenstein E. Cat. Rev. Sci. Eng. ,1984 , 26 (1 ), 1 —58
[本文引用: 1]
[8]
Park S. Hwang H. J. Moon J. Catal. Lett. , 2003 , 87 (3/4 ), 219 —223
[本文引用: 1]
[9]
Sohn J. M. Kim M. R. Woo S. I. Catal. Today , 2003 , 83 (1—4 ), 289 —297
[本文引用: 1]
[10]
Cheng J. Wang H. Hao Z. Wang S. Catal. Commun. , 2008 , 9 (5 ), 690 —695
[本文引用: 1]
[11]
Niu X. Li H. Liu G. J. Mol. Catal. A: Chem. , 2005 , 232 (1/2 ), 89 —93
[本文引用: 1]
[12]
McCauley R. A. J. Appl. Phys. , 1980 , 51 (1 ), 290 —294
[本文引用: 1]
[13]
Hornés A. Hungría A. B. Bera P. Cámara A. L. Fernández-García M. Martínez-Arias A. Barrio L. Estrella M. Zhou G. Fonseca J. J. Hanson J. C. Rodriguez J. A. J. Am. Chem. Soc. , 2010 , 132 (1 ), 34 —35
[本文引用: 1]
[14]
Chmielarz L. Kus'trowski P. Rafalska-Łasocha A. Dziembaj R. Thermochim. Acta , 2002 , 395 (1/2 ), 225 —236
[本文引用: 1]
[15]
Jiang Z. Yu J. Cheng J. Xiao T. M. O. Jones, Hao Z. , Edwards P. P. Fuel Process. Technol. , 2010 , 91 (1 ), 97 —102
[本文引用: 1]
[16]
Lee J. G. Lee C. M. Park M. Shul Y. G. RSC Adv. , 2013 , 29 (3 ), 11816 —11822
[本文引用: 1]
[17]
Liu J. Zhao Z. Wang J. Xu C. Duan A. Jiang G. Yang Q. Appl. Catal. B: Environ. , 2008 , 84 (1/2 ), 185 —195
[本文引用: 1]
[18]
Yao H. C. Yao Y. F. Y. J. Catal. , 1984 , 86 (2 ), 254 —265
[本文引用: 1]
[19]
Scirè S. Minicò S. Crisafulli C. Satriano C. Pistone A. Appl. Catal. B: Environ. , 2003 , 40 (1 ), 43 —49
[本文引用: 1]
[20]
Shek C. H. Lin G. M. , Lai J. K. L. , Nanostruct. Mater. , 1999 , 11 (7 ), 831 —835
[本文引用: 1]
[21]
Du Z. Dunlap R. A. Obrovac M. N. J. Alloys Compd. , 2014 , 617 , 271 —276
[本文引用: 1]
[22]
Zhang-Steenwinkel Y. Beckers J. Bliek A. Appl. Catal. A: Gen. , 2002 , 235 (1/2 ), 79 —92
[本文引用: 1]
[23]
Yokoi Y. Uchida H. Catal. Today , 1998 , 42 (1 ), 167 —174
[24]
Lin X. M. Zhu L. L. Han J. Liu X. M. Chem. J. Chinese Universities , 2015 , 36 (1 ), 61 —66
[本文引用: 1]
(林晓敏 , 朱丽丽 , 韩健 , 刘晓梅 . 高等学校化学学报 2015 , 36 (1 ), 61 —66 )
[本文引用: 1]
[25]
Xu X. Zhang R. Zeng X. Han X. Li Y. Liu Y. Wang X. Chem. Catal. Chem. , 2013 , 5 (7 ), 2025 —2036
[本文引用: 1]
[26]
Sujatha Lekshmy S. Joy K. J. Mater. Sci. Mater. Electron. , 2014 , 25 (4 ), 1664 —1672
[本文引用: 1]
[27]
Ansell R. O. Dickinson T. Povey A. F. Sherwood P. M. A. J. Electron. Spectrosc. Relat. Phenom. , 1977 , 11 (3 ), 301 —313
[本文引用: 1]
[28]
Sun M. Su Y. Du C. Zhao Q. Liu Z. RSC Adv. , 2014 , 4 (58 ), 30820 —30827
[本文引用: 1]
[29]
Choudhary T. V. Banerjee S. Choudhary V. R. Appl. Catal. A: Gen. , 2002 , 234 (1/2 ), 1 —23
[本文引用: 1]
[30]
Zhang C. Grass M. E. McDaniel A. H. DeCaluwe S. C. El Gabaly F. Liu Z. McCarty K. F. Farrow R. L. Linne M. A. Hussain Z. Nature Materials , 2010 , 9 (11 ), 944 —949
[本文引用: 1]
[31]
Popescu A. M. Chem. Res. Chinese Universities , 2014 , 30 (5 ), 800 —805
[本文引用: 1]
[32]
Sutthiumporn K. Kawi S. Int. J. Hydrogen Energy , 2011 , 36 (22 ), 14435 —14446
[本文引用: 1]
[33]
Liu Q. Y. He S. G. Chem. J. Chinese Universities , 2014 , 35 (4 ), 665 —688
[本文引用: 1]
(刘清宇 , 何圣贵 . 高等学校化学学报 ,2014 , 35 (4 ), 665 —688 )
[本文引用: 1]
[34]
Ren C. Zhou L. Duan Y. Chen Y. J. Rare Earths , 2012 , 30 (11 ), 1106 —1111
[本文引用: 1]
[35]
Deng J. G. He S. N. Xie S. H. Yang H. G. Liu Y. X. Dai H. X. Chem. J. Chinese Universities , 2014 , 35 (6 ), 1119 —1129
[本文引用: 1]
(邓积光 , 何胜男 , 谢少华 , 杨黄根 , 刘雨溪 , 戴洪兴 . 高等学校化学学报 ,2014 , 35 (6 ), 1119 —1129 )
[本文引用: 1]
1
2004
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
2013
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
1995
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
1984
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
2003
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
2003
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
2008
... 天然气(主要成分为甲烷)作为一种清洁能源燃料, 其应用和研究领域已得到迅速扩展和深入[1 ] . 作为燃料燃烧, 天然气采用的方式多为传统火焰燃烧, 火焰燃烧温度可高达2000 ℃, 能使空气中大量的N2 转化为NOx [2 ] , 将天然气燃烧温度控制在1500 ℃以下[3 ~7 ] , 有效地避免NOx 2 Sn2 O7 , Ln2 Zr2 O7 等未掺杂的烧绿石型氧化物作为甲烷燃烧催化剂[8 ,9 ] , 但其催化效果不够理想. 2008年Cheng等[10 ] 考察了利用共沉淀法制备的掺杂钴的烧绿石型氧化物La2 Sn1- x x 7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ
1
2005
... 样品a在2θ 为29.0°, 36.6°和57.1°的衍射峰对应烧绿石的晶面(222), (331)和(622)的特征衍射峰, 且未检测到其它物相特征峰, 表明物相为单一的La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的(111), (200), (220)和(311)晶面所对应的特征衍射峰, 说明此样品由La2 Sn1.7 Co0.3 O7- δ 2 2种物相组成. 样品c中同样也出现了La2 Sn1.7 Co0.3 O7- δ 2 2种物相, 但值得注意的是, 机械混合样品c的CeO2 衍射峰强度比逆负载催化剂b的更强, 说明逆负载催化剂b中CeO2 在La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的负载对La2 Sn1.7 Co0.3 O7- δ 表1 . 由表1 可见, 逆负载催化剂b与单一烧绿石型催化剂a相比, La2 Sn1.7 Co0.3 O7- δ 2 的负载能够有效阻止La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的影响弱于烧绿石逆负载型催化剂. 通过公式[11 ] : (2ω )2 cos2 θ =4/π 2 (λ /Dhkl )2 +32< ε 2 h kl 2 θ , 计算相关催化剂中烧绿石晶格的扭曲程度, 其中Dhkl 是晶面间距; λ 为X射线波长(λ =0.15406 nm); θ 为晶面(hkl )的衍射角; 2ω 为校正的衍射峰半高宽(FWHM); < ε 2 h kl 1/2 为晶面扭曲度, 计算结果列于表1 . 逆负载催化剂b中的烧绿石晶体结构发生了较大程度的扭曲, 表明CeO2 对La2 Sn1.7 Co0.3 O7- δ [12 ] . ...
1
1980
... 样品a在2θ 为29.0°, 36.6°和57.1°的衍射峰对应烧绿石的晶面(222), (331)和(622)的特征衍射峰, 且未检测到其它物相特征峰, 表明物相为单一的La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的(111), (200), (220)和(311)晶面所对应的特征衍射峰, 说明此样品由La2 Sn1.7 Co0.3 O7- δ 2 2种物相组成. 样品c中同样也出现了La2 Sn1.7 Co0.3 O7- δ 2 2种物相, 但值得注意的是, 机械混合样品c的CeO2 衍射峰强度比逆负载催化剂b的更强, 说明逆负载催化剂b中CeO2 在La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的负载对La2 Sn1.7 Co0.3 O7- δ 表1 . 由表1 可见, 逆负载催化剂b与单一烧绿石型催化剂a相比, La2 Sn1.7 Co0.3 O7- δ 2 的负载能够有效阻止La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 的影响弱于烧绿石逆负载型催化剂. 通过公式[11 ] : (2ω )2 cos2 θ =4/π 2 (λ /Dhkl )2 +32< ε 2 h kl 2 θ , 计算相关催化剂中烧绿石晶格的扭曲程度, 其中Dhkl 是晶面间距; λ 为X射线波长(λ =0.15406 nm); θ 为晶面(hkl )的衍射角; 2ω 为校正的衍射峰半高宽(FWHM); < ε 2 h kl 1/2 为晶面扭曲度, 计算结果列于表1 . 逆负载催化剂b中的烧绿石晶体结构发生了较大程度的扭曲, 表明CeO2 对La2 Sn1.7 Co0.3 O7- δ [12 ] . ...
1
2010
... 图4 为催化剂样品的透射电子显微镜(TEM)、 高分辨透射电子显微镜(HRTEM)和原子力显微镜(AFM)照片. 图4 (A)~(C)分别是单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 图4 (A), (C)可见, 催化剂大部分颗粒聚集在一起, 表面发生严重烧结, 颗粒分布在40~70 nm之间, 由图4 (B)可观察到样品的几个晶面均为具有烧绿石复合氧化物La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 图4 (D)~(F)为CeO2 /La2 Sn1.7 Co0.3 O7- δ 图4 (D)中呈现出大小不同且相互交错的颗粒形貌, 由图4 (E)的HRTEM照片可确定, 晶面间距为0.31 nm的晶面, 对应烧绿石型晶体La2 Sn1.7 Co0.3 O7- δ 2 的(220)晶面, 故可确定具有较小粒径的颗粒为CeO2 粒子, 具有较大粒径的颗粒为La2 Sn1.7 Co0.3 O7- δ 2 粒子负载到较大的La2 Sn1.7 Co0.3 O7- δ 2 /CuO催化剂的颗粒形貌相似[13 ] . 根据TEM照片可测量出CeO2 颗粒尺寸在5~11 nm之间, 烧绿石样品粒径在20~50 nm之间, 较单一La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载烧绿石La2 Sn1.7 Co0.3 O7- δ
1
2002
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
2010
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
2013
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
2008
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
1984
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
2003
... 图5 给出了相关催化剂的H2 -TPR曲线. 由图5 可见, 单一La2 Sn1.7 Co0.3 O7- δ 3+ 到Co2+ 及Co2+ 到Co0 的还原[14 ,15 ] , 第三个宽且大的峰在623 ℃左右, 归属为La2 Sn1.7 Co0.3 O7- δ 4+ 到Sn2+ 的还原[16 ] . 与单一烧绿石催化剂相比, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 3+ 和Co2+ 的还原峰位置向低温方向有明显偏移, 还原峰最高位置分别为382和440 ℃, 明显低于单一烧绿石催化剂相应还原峰的温度, 说明CeO2 对烧绿石结构中Co的还原有促进作用, 有利于降低钴离子的还原温度[17 ] ; 此外, 逆负载催化剂中Sn4+ 到Sn2+ 的还原温度也明显降低, 还原峰位置由623 ℃降到599 ℃, 降低了24 ℃, 可见CeO2 对烧绿石La2 Sn1.7 Co0.3 O7- δ 4+ 的还原反应有较大的促进作用, 表明CeO2 和La2 Sn1.7 Co0.3 O7- δ 2 的负载可以有效降低La2 Sn1.7 Co0.3 O7- δ 2 起到降低烧绿石结构中Sn—O和Co—O键能的作用, 进而促进催化剂中活性氧从晶格内部向材料表面的转移, 提高烧绿石氧化物结构中氧的移动性. 由图5 曲线c 发现CeO2 在553 ℃处出现了1个还原峰, 在800 ℃以后出现了1个不完整的还原峰. 普遍认为[18 ,19 ] 500 ℃左右的还原峰反映了CeO2 表面Ce4+ 到Ce3+ 的还原, 800 ℃左右的还原峰反映了CeO2 体相Ce4+ 到Ce3+ 的还原, 所以, 本文的实验结果与CeO2 的H2 -TPR的还原温度基本符合. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的特征还原峰, 即CeO2 表面Ce4+ 到Ce3+ 的还原, 但逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的第一个特征还原峰比单一CeO2 的还原温度降低了20 ℃, 说明烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 有一定的分散作用, CeO2 很好地负载在烧绿石型氧化物上. 此外, CeO2 在800 ℃以后出现的不完整还原峰由于与烧绿石La2 Sn1.7 Co0.3 O7- δ 2+ 到Sn0 的还原峰相重叠, 因此难以辨别. ...
1
1999
... 由图6 可知, 2个样品中均有2种亚谱, 化学位移均在零速度附近, 且均为四极分裂双峰, 可判定亚谱中的锡均为Sn4+ , 故2种催化剂中Sn的价态主要以+4价存在. 另外, 根据文献[20 ]报道的SnO2 穆斯堡尔谱分析, 将催化剂中的亚谱1归属为界面相的Sn4+ , 亚谱2归属为结晶相的Sn4+ . 表3 列出了各样品的穆斯堡尔参数, 单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 4+ 含量降低, 结晶相Sn4+ 含量升高, 这可能正是甲烷燃烧催化活性提高的原因之一. 此外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 相互作用, 致使Sn核处的s 电子密度变小, 从而导致产生的IS值变大; 另外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的影响, 致使烧绿石晶体结构发生一定程度的畸变[21 ] , 促进甲烷燃烧催化活性的提高. ...
1
2014
... 由图6 可知, 2个样品中均有2种亚谱, 化学位移均在零速度附近, 且均为四极分裂双峰, 可判定亚谱中的锡均为Sn4+ , 故2种催化剂中Sn的价态主要以+4价存在. 另外, 根据文献[20 ]报道的SnO2 穆斯堡尔谱分析, 将催化剂中的亚谱1归属为界面相的Sn4+ , 亚谱2归属为结晶相的Sn4+ . 表3 列出了各样品的穆斯堡尔参数, 单一烧绿石型催化剂La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 4+ 含量降低, 结晶相Sn4+ 含量升高, 这可能正是甲烷燃烧催化活性提高的原因之一. 此外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 相互作用, 致使Sn核处的s 电子密度变小, 从而导致产生的IS值变大; 另外, 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 的影响, 致使烧绿石晶体结构发生一定程度的畸变[21 ] , 促进甲烷燃烧催化活性的提高. ...
1
2002
... 图7 为催化剂的X射线光电子能谱. 用C1 s 图7 (A)给出单一烧绿石催化剂和逆负载催化剂的O1 s 1 s [22 ~24 ] , 通常将528~530 eV低结合能处的峰归属为晶格氧, 531~533 eV高结合能处的峰归属为吸附氧. 单一La2 Sn1.7 Co0.3 O7- δ [25 ] , 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 使La2 Sn1.7 Co0.3 O7- δ 2 与La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 没有明显影响到催化剂的吸附氧结合能变化, 但却使催化剂晶格氧的结合能发生了较明显的偏移. 表4 列出了催化剂样品O1 s 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载到La2 Sn1.7 Co0.3 O7- δ
1
2015
... 图7 为催化剂的X射线光电子能谱. 用C1 s 图7 (A)给出单一烧绿石催化剂和逆负载催化剂的O1 s 1 s [22 ~24 ] , 通常将528~530 eV低结合能处的峰归属为晶格氧, 531~533 eV高结合能处的峰归属为吸附氧. 单一La2 Sn1.7 Co0.3 O7- δ [25 ] , 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 使La2 Sn1.7 Co0.3 O7- δ 2 与La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 没有明显影响到催化剂的吸附氧结合能变化, 但却使催化剂晶格氧的结合能发生了较明显的偏移. 表4 列出了催化剂样品O1 s 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载到La2 Sn1.7 Co0.3 O7- δ
1
2015
... 图7 为催化剂的X射线光电子能谱. 用C1 s 图7 (A)给出单一烧绿石催化剂和逆负载催化剂的O1 s 1 s [22 ~24 ] , 通常将528~530 eV低结合能处的峰归属为晶格氧, 531~533 eV高结合能处的峰归属为吸附氧. 单一La2 Sn1.7 Co0.3 O7- δ [25 ] , 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 使La2 Sn1.7 Co0.3 O7- δ 2 与La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 没有明显影响到催化剂的吸附氧结合能变化, 但却使催化剂晶格氧的结合能发生了较明显的偏移. 表4 列出了催化剂样品O1 s 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载到La2 Sn1.7 Co0.3 O7- δ
1
2013
... 图7 为催化剂的X射线光电子能谱. 用C1 s 图7 (A)给出单一烧绿石催化剂和逆负载催化剂的O1 s 1 s [22 ~24 ] , 通常将528~530 eV低结合能处的峰归属为晶格氧, 531~533 eV高结合能处的峰归属为吸附氧. 单一La2 Sn1.7 Co0.3 O7- δ [25 ] , 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 使La2 Sn1.7 Co0.3 O7- δ 2 与La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 Sn1.7 Co0.3 O7- δ 2 没有明显影响到催化剂的吸附氧结合能变化, 但却使催化剂晶格氧的结合能发生了较明显的偏移. 表4 列出了催化剂样品O1 s 2 /La2 Sn1.7 Co0.3 O7- δ 2 负载到La2 Sn1.7 Co0.3 O7- δ
1
2014
... 图7 (B)为催化剂中Sn3 d 2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 4+ 结合能峰[26 ,27 ] , 故可确定样品中Sn价态主要为+4价, 与穆斯堡尔谱测试结果相符. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 与烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 改变了Sn周围的电子云密度, 使Sn周边的化学环境发生变化; 而且CeO2 的负载降低了催化剂中Sn3 d [28 ] , 从而使逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 表4 可知, La2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 2 /La2 Sn1.7 Co0.3 O7- δ 3 d 2 的负载对烧绿石结构中S n 3 d 3 2 n 3 d 5 2 3 d 图7 (C), L a 3 d 3 2 a 3 d 5 2 3+ 的结合能没有受到CeO2 的影响. 由于添加的钴含量过少, 在XPS测试中钴的结合能较微弱, 因此, 不能明确确定CeO2 的影响. ...
1
1977
... 图7 (B)为催化剂中Sn3 d 2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 4+ 结合能峰[26 ,27 ] , 故可确定样品中Sn价态主要为+4价, 与穆斯堡尔谱测试结果相符. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 与烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 改变了Sn周围的电子云密度, 使Sn周边的化学环境发生变化; 而且CeO2 的负载降低了催化剂中Sn3 d [28 ] , 从而使逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 表4 可知, La2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 2 /La2 Sn1.7 Co0.3 O7- δ 3 d 2 的负载对烧绿石结构中S n 3 d 3 2 n 3 d 5 2 3 d 图7 (C), L a 3 d 3 2 a 3 d 5 2 3+ 的结合能没有受到CeO2 的影响. 由于添加的钴含量过少, 在XPS测试中钴的结合能较微弱, 因此, 不能明确确定CeO2 的影响. ...
1
2014
... 图7 (B)为催化剂中Sn3 d 2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 4+ 结合能峰[26 ,27 ] , 故可确定样品中Sn价态主要为+4价, 与穆斯堡尔谱测试结果相符. 逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 2 /La2 Sn1.7 Co0.3 O7- δ 2 与烧绿石型氧化物La2 Sn1.7 Co0.3 O7- δ 2 改变了Sn周围的电子云密度, 使Sn周边的化学环境发生变化; 而且CeO2 的负载降低了催化剂中Sn3 d [28 ] , 从而使逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ 表4 可知, La2 Sn1.7 Co0.3 O7- δ n 3 d 3 2 n 3 d 5 2 2 /La2 Sn1.7 Co0.3 O7- δ 3 d 2 的负载对烧绿石结构中S n 3 d 3 2 n 3 d 5 2 3 d 图7 (C), L a 3 d 3 2 a 3 d 5 2 3+ 的结合能没有受到CeO2 的影响. 由于添加的钴含量过少, 在XPS测试中钴的结合能较微弱, 因此, 不能明确确定CeO2 的影响. ...
1
2002
... 在没有添加催化剂的情况下, 甲烷燃烧反应的机理为自由基反应机理, 自由基剧烈运动, 会导致甲烷反应温度的急剧上升, 高达2000 ℃以上. 加入催化剂后, 甲烷燃烧反应机理变得十分复杂, 此时, 不但会发生自由基反应, 而且还会发生表面氧化反应. 对于耐高温性较好的烧绿石型复合氧化物催化剂, 催化剂表面吸附氧和晶格氧是影响催化剂活性的主要因素, 通常将甲烷催化燃烧反应分为“面上反应(suprafacial)”和“面内反应(intrafacial)”两种反应机制[29 ] , 在温度较低时一般是吸附氧起主要作用, 属于“面上反应”机制, 在温度较高时一般是晶格氧起主要作用, 属于“面内反应”机制. 用耐高温性较好的烧绿石复合氧化物La2 Sn1.7 Co0.3 O7- δ
1
2010
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2014
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2011
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2014
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2014
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2012
... 式中, Vo代表氧空穴, 此时O2- 主要与催化剂中的晶格氧有关[30 ,31 ] , 式(1)为Sn4+ 的还原反应, 此过程中催化剂先与晶格氧中可迁移的活性氧作用, 转化甲烷并生成氧空穴; 式(2)为Sn2+ 的氧化反应, 新鲜的氧气填充到氧空穴, 与催化剂接触, 将催化剂中Sn2+ 氧化为Sn4+ , 如此循环, 以达到甲烷催化转化的目的. 由反应机理可知, 催化剂表面氧空穴Vo和氧种类O2- 的多少, 对催化剂的催化活性起重要作用. 上述XPS表征显示出逆负载催化剂结构发生畸变, 晶格氧结合能降低, 这可能对氧空穴的产生有影响[32 ] , 氧空穴能吸收大部分氧并将之转化成 O 2 2 - O 2 - - 等氧离子[33 ] , 进而促进催化剂表面氧种类的增多[34 ] , 因此, 逆负载催化剂具有良好催化活性很有可能与表面氧空穴和氧种类的增多有关. 对La2 Sn1.7 Co0.3 O7- δ 2 应用于甲烷燃烧反应时, 反应机理则稍有一些差别, 其反应过程可写为 ...
1
2014
... 式中, O 2 ( Ce O 2 ) 2 自身释放出来的O2 . 由于CeO2 具有较强的储放氧功能[35 ] , 将CeO2 应用于甲烷催化燃烧反应, CeO2 中的部分氧可以释放出来用于氧化甲烷, 而自身变成Ce2 O3 , Ce2 O3 在反应气提供的O2 过剩的条件下又恢复成CeO2 . 因此, 我们认为逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ
1
2014
... 式中, O 2 ( Ce O 2 ) 2 自身释放出来的O2 . 由于CeO2 具有较强的储放氧功能[35 ] , 将CeO2 应用于甲烷催化燃烧反应, CeO2 中的部分氧可以释放出来用于氧化甲烷, 而自身变成Ce2 O3 , Ce2 O3 在反应气提供的O2 过剩的条件下又恢复成CeO2 . 因此, 我们认为逆负载催化剂CeO2 /La2 Sn1.7 Co0.3 O7- δ


 , 白雅琴
, 白雅琴




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}