Chem. J. Chinese Universities ›› 2016, Vol. 37 ›› Issue (4): 674.doi: 10.7503/cjcu20150921
• Organic Chemistry • Previous Articles Next Articles
Theoretical Studies on Tetragonal, Monoclinic and Orthorhombic Distortions of Germanium Nitride Polymorphs†
CANG Yuping, CHEN Dong*( ), YANG Fan, YANG Huiming
), YANG Fan, YANG Huiming
- College of Physics and Electronic Engineering, Xinyang Normal University, Xinyang 464000, China
-
Received:2015-11-29Online:2016-04-10Published:2016-03-17 -
Contact:CHEN Dong E-mail:chchendong2010@163.com -
Supported by:† Supported by the National Natural Science Foundation of China(Nos.61475132, 11304141, 11475143, 61501392) and the National Training Programs of Innovation and Entrepreneurship for Undergraduates, China(No.201510477001)
CLC Number:
TrendMD:
Cite this article
CANG Yuping, CHEN Dong, YANG Fan, YANG Huiming. Theoretical Studies on Tetragonal, Monoclinic and Orthorhombic Distortions of Germanium Nitride Polymorphs†[J]. Chem. J. Chinese Universities, 2016, 37(4): 674.
share this article
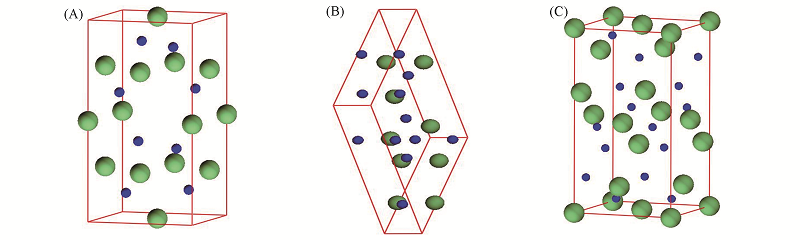
Fig.1 Crystal structures of tetragonal(A), monoclinic(B) and orthorhombic(C) Ge3N4 phases ^The small and big balls represent the N and Ge atoms, respectively.
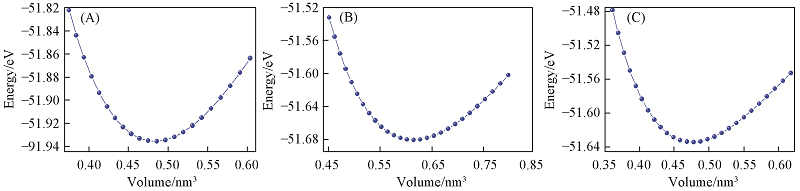
Fig.2 Total energy as a function of volume for t-Ge3N4(A), m-Ge3N4(B) and o-Ge3N4(C)
| Species | t-Ge3N4 | m-Ge3N4 | o-Ge3N4 | c-Ge3N4 | ||||
|---|---|---|---|---|---|---|---|---|
| 0 GPa | 20 GPa | 0 GPa | 20 GPa | 0 GPa | 20 GPa | Calcd. | Expt. | |
| a | 0.4410 | 0.4240 | 1.0215 | 0.9735 | 0.5256 | 0.5104 | a=0.8212 | a=0.8173[ |
| b | 0.3103 | 0.2987 | 1.0074 | 0.9901 | B=221.8 | B=242.0[ | ||
| c | 0.8835 | 0.8524 | 0.7928 | 0.7789 | 0.5319 | 0.5119 | G=170.9 | G=176.2[ |
| ΔH | -24.16 | -14.06 | -24.34 | -13.77 | -23.09 | -14.68 | C11=378.8 | C11=395.1[ |
| B | 146.8 | 206.8 | 124.1 | 189.1 | 203.4 | 122.1 | C12=147.9 | C12=165.4[ |
| G | 86.8 | 85.4 | 72.8 | 65.1 | 296.1 | 132.5 | C44=223.7 | C44=234.5[ |
| C11 | 200.4 | 249.6 | 175.2 | 236.8 | 300.1 | 411.6 | a=0.8210[ | |
| C12 | 125.5 | 200.5 | 31.3 | 80.3 | 170.8 | 268.5 | B=296[ | |
| C13 | 109.6 | 167.7 | 106.5 | 169.7 | 139.7 | 238.4 | B=268.6[ | |
| C22 | 337.9 | 459.8 | 418.2 | 520.7 | a=0.8168[ | |||
| C23 | 41.1 | 79.9 | 99.0 | 171.9 | a=0.8211[ | |||
| C25 | 9.8 | 8.9 | a=0.8120[ | |||||
| C33 | 232.2 | 286.4 | 276.7 | 400.3 | 311.5 | 399.1 | ||
| C35 | 19.9 | 35.2 | 151.8 | |||||
| C44 | 124.8 | 140.5 | 54.6 | 46.0 | 140.8 | |||
| C46 | 22.4 | 22.2 | ||||||
| C55 | 90.4 | 104.2 | 143.9 | 164.5 | ||||
| C66 | 147.6 | 169.9 | 62.6 | 30.8 | 141.1 | 161.8 | | |
Table 1 Lattice constants a, b, c (nm), formation enthalpy ΔH (eV), bulk modulus B, shear modulus G and elastic constants Cij(GPa) of Ge3N4
| Species | t-Ge3N4 | m-Ge3N4 | o-Ge3N4 | c-Ge3N4 | ||||
|---|---|---|---|---|---|---|---|---|
| 0 GPa | 20 GPa | 0 GPa | 20 GPa | 0 GPa | 20 GPa | Calcd. | Expt. | |
| a | 0.4410 | 0.4240 | 1.0215 | 0.9735 | 0.5256 | 0.5104 | a=0.8212 | a=0.8173[ |
| b | 0.3103 | 0.2987 | 1.0074 | 0.9901 | B=221.8 | B=242.0[ | ||
| c | 0.8835 | 0.8524 | 0.7928 | 0.7789 | 0.5319 | 0.5119 | G=170.9 | G=176.2[ |
| ΔH | -24.16 | -14.06 | -24.34 | -13.77 | -23.09 | -14.68 | C11=378.8 | C11=395.1[ |
| B | 146.8 | 206.8 | 124.1 | 189.1 | 203.4 | 122.1 | C12=147.9 | C12=165.4[ |
| G | 86.8 | 85.4 | 72.8 | 65.1 | 296.1 | 132.5 | C44=223.7 | C44=234.5[ |
| C11 | 200.4 | 249.6 | 175.2 | 236.8 | 300.1 | 411.6 | a=0.8210[ | |
| C12 | 125.5 | 200.5 | 31.3 | 80.3 | 170.8 | 268.5 | B=296[ | |
| C13 | 109.6 | 167.7 | 106.5 | 169.7 | 139.7 | 238.4 | B=268.6[ | |
| C22 | 337.9 | 459.8 | 418.2 | 520.7 | a=0.8168[ | |||
| C23 | 41.1 | 79.9 | 99.0 | 171.9 | a=0.8211[ | |||
| C25 | 9.8 | 8.9 | a=0.8120[ | |||||
| C33 | 232.2 | 286.4 | 276.7 | 400.3 | 311.5 | 399.1 | ||
| C35 | 19.9 | 35.2 | 151.8 | |||||
| C44 | 124.8 | 140.5 | 54.6 | 46.0 | 140.8 | |||
| C46 | 22.4 | 22.2 | ||||||
| C55 | 90.4 | 104.2 | 143.9 | 164.5 | ||||
| C66 | 147.6 | 169.9 | 62.6 | 30.8 | 141.1 | 161.8 | | |
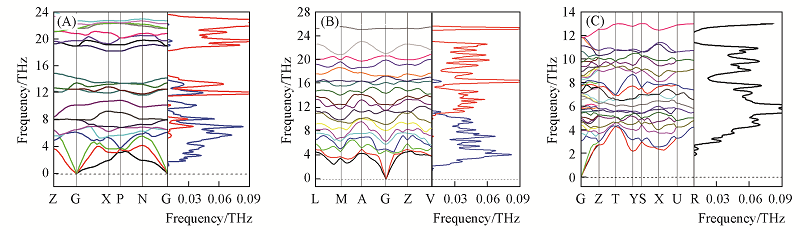
Fig.3 Phonon dispersions and phonon density of states for t-Ge3N4(A), m-Ge3N4(B) and o-Ge3N4(C)^ The blue and red lines are the DOS of N and Ge, respectively.
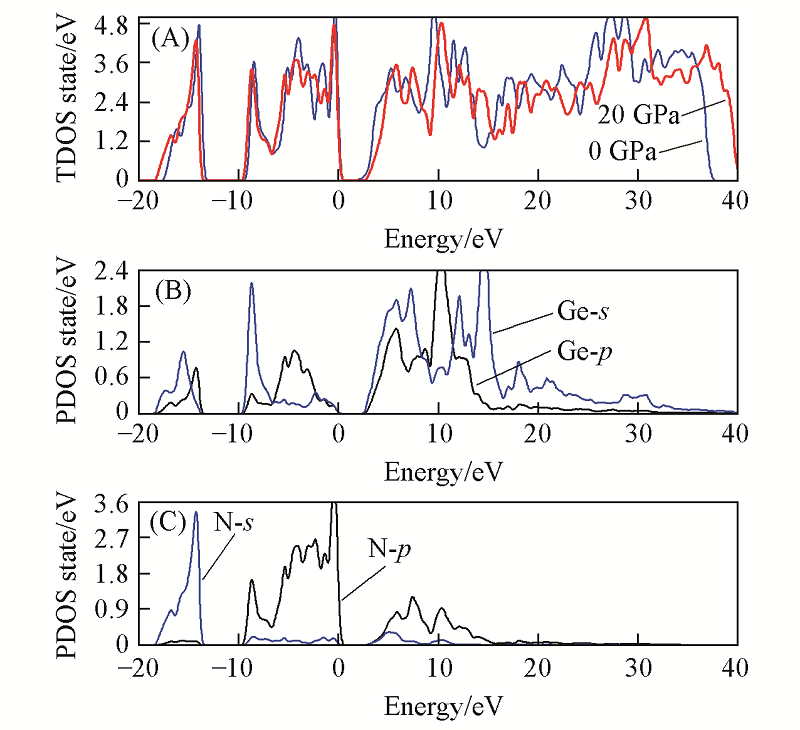
Fig.4 Total(A) and partial density of states for Ge(B), N(C) atoms of t-Ge3N4
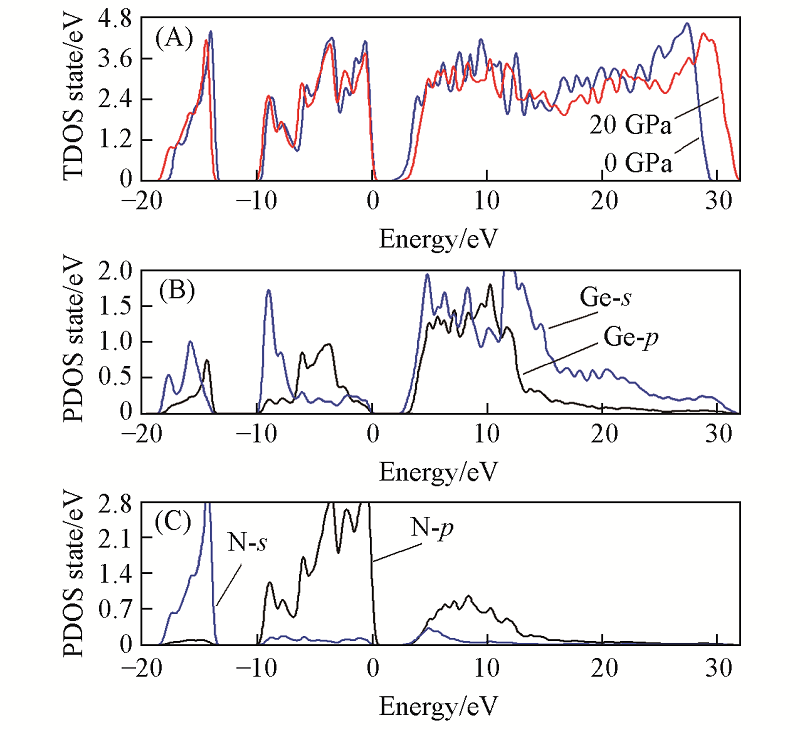
Fig.5 Total(A) and partial density of states for Ge(B), N(C) atoms of m-Ge3N4
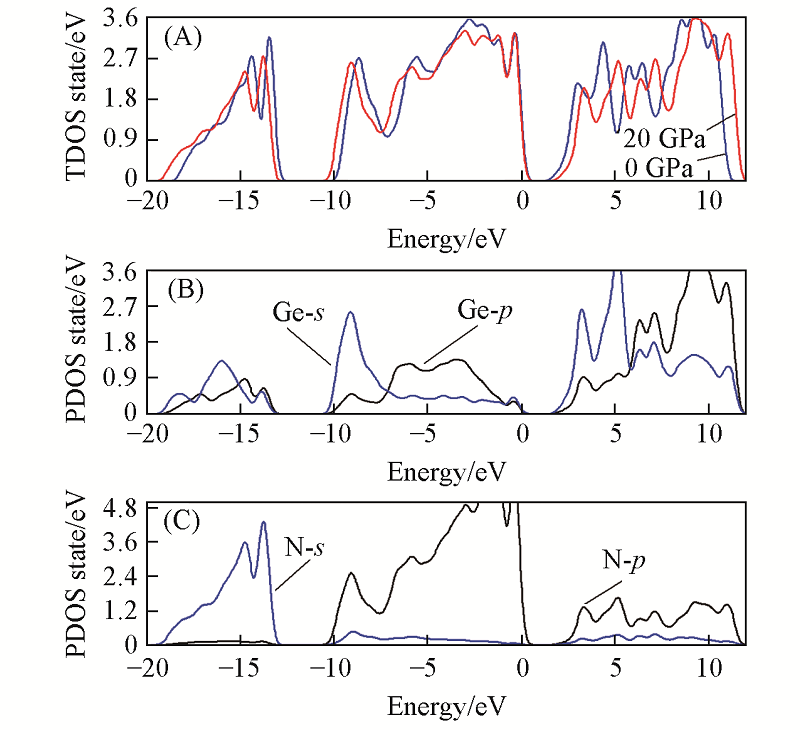
Fig.6 Total(A) and partial density of states for Ge(B), N(C) atoms of o-Ge3N4
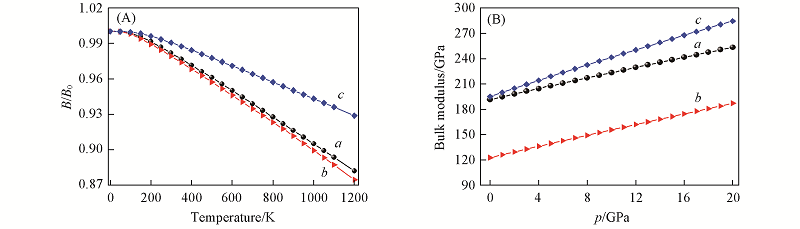
Fig.7 Temperature dependences of the normalized bulk moduli B/B0(A) and pressure dependences of the bulk moduli B(B) for the new Ge3N4 phases^a. t-Ge3N4; b. m-Ge3N4; c. o-Ge3N4.
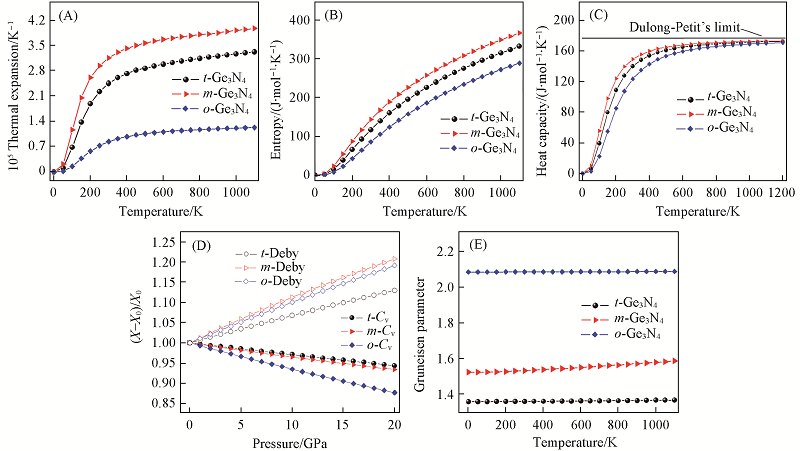
Fig.8 Variations of the volume thermal expansion α(V)(A), entropy S(B), heat capacity Cv(C), normalized Debye temperature θD and heat capacity Cv (D) and Grüneisen parameter γ(E) for the three Ge3N4 compounds
| [1] | Lu X., La P., Guo X., Wei Y., Nan X., He L., J. Phys. Chem. Lett., 2013, 4(11), 1878—1881 |
| [2] | Fu R. B., Yang L. Q., Feng L. Y., Guo W., Chem. J. Chinese Universities, 2014, 35(4), 825—830 |
| (付融冰, 杨兰琴, 冯雷雨, 郭伟. 高等学校化学学报, 2014, 35(4), 825—830) | |
| [3] | Hart J. N., Allan N. L., Claeyssens F., Phys. Rev. B, 2011, 84(24), 245209 |
| [4] | Yang H. M., Xu B., Guo X. F., Wen Z. X., Fan X. H., Tian B., Chem. J. Chinese Universities, 2013, 34(4), 970—974 |
| (杨红梅, 许斌, 郭晓斐, 温振兴, 范小红, 田彬. 高等学校化学学报, 2013, 34(4), 970—974) | |
| [5] | Salamat A., Hector A. L., Kroll P., McMillan P. F., Coordin. Chem. Rev., 2013, 257(13/14), 2063—2072 |
| [6] | Ding Y. C., Chen M., Wu W. J., J. Theor. Comput. Chem., 2015, 14(4), 1550024 |
| [7] | Boyko T. D., Hunt A., Zerr A., Moewes A., Phys. Rev. Lett., 2013, 111(9), 097402 |
| [8] | Deb S. K., Dong J., Hubert H., McMillan P. F., Sankey O. F., Solid State Commun., 2000, 114(3), 137—142 |
| [9] | Duan Y. H., Zhang K. M., Xie X. D., Acta Phys. Sinica, 1996, 45(3), 512—517 |
| (段玉华, 张开明, 谢希德. 物理学报, 1996, 45(3), 512—517) | |
| [10] | Serghiou G., Miehe G., Tschauner O., Zerr A., Boehler R., J. Chem. Phys., 1999, 111(10), 4659—4662 |
| [11] | Leinenweber K., O’Keeffe M., Somayazulu M. S., Hubert H., McMillan P. F., Wolf G. H., Chem. Eur. J., 1999, 5(10), 3076—3078 |
| [12] | Yang M., Wang S. J., Feng Y. P., Peng G. W., Sun Y. Y., J. Appl. Phys., 2007, 102(1), 013507 |
| [13] | Chu L. H., Kozhevnikov A., Schulthess T. C., Cheng H. P., J. Chem. Phys., 2014, 141(4), 044709 |
| [14] | He H. L., Sekine T., Kobayashi T., Kimoto K., J. Appl. Phys., 2001, 90(9), 4403—4406 |
| [15] | Wang Z., Zhao Y., Schiferl D., Qian J., Downs R. T., Mao H. K., Sekine T., J. Phys. Chem. B, 2013, 107(51), 14151—14153 |
| [16] | Dong J. J., Sankey O. F., Deb S. K., Wolf G., McMillan P. F., Phys. Rev. B, 2000, 61(18), 11979—11992 |
| [17] | Gao S. P., Cai G. H., Xu Y., Comput. Mater. Sci., 2013, 67, 292—295 |
| [18] | Lü T. Y., Zheng J. C., Chem. Phys. Lett., 2010, 501(1—3), 47—53 |
| [19] | Wang H., Chen Y., Kaneta Y., Iwata S., J. Phys.: Condens. Matter., 2006, 18(47), 10663—10676 |
| [20] | Ching W. Y., Mo S. D., Ouyang L. Z., Phys. Rev. B, 2001, 63(24), 245110 |
| [21] | Horvath-Bordon E., Riedel R., Zerr A., McMillan P. F., Auffermann G., Prots Y., Bronger W., Kniep R., Kroll P., Chem. Soc. Rev., 2006, 35(10), 987—1014 |
| [22] | Soignard E., McMillan P. F., Hejny C., Leinenweber K., J. Solid State Chem., 2004, 177(1), 299—311 |
| [23] | McMillan P. F., Deb S. K., Dong J. J., J. Raman Spectrosc., 2003, 34(7/8), 567—577 |
| [24] | Luo Y., Cang Y., Chen D., Comput. Condens. Matter., 2014, 1, 1—7 |
| [25] | Lowther J. E., Phys. Rev. B, 2000, 62(1), 5—8 |
| [26] | Cui L., Hu M., Wang Q., Xu B., Yu D., Liu Z., He J., J. Solid State Chem., 2015, 228, 20—26 |
| [27] | Kohn W., Sham L. J., Phys. Rev., 1965, 140(4A), A1133—A1138 |
| [28] | Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett., 1996, 77(18), 3865—3868 |
| [29] | Monkhorst H. J., Pack J. D., Phys. Rev. B, 1976, 13(12), 5188—5192 |
| [30] | Ackland G. J., Warren M. C., Clark S. J., J. Phys.: Condens. Matter., 1997, 9(37), 7861—7872 |
| [31] | Shanno D. F., Kettler P. C., Math. Comput., 1970, 24(1—3), 657—664 |
| [32] | Otero-de-la-Roza A., Luańa V., Comput. Phys. Commun., 2011, 182(8), 1708—1720 |
| [33] | Otero-de-la-Roza A., Abbasi-Pérez D., Luańa V., Comput. Phys. Commun., 2011, 182(10), 2232—2248 |
| [34] | Dong J. J., Deslippe J., Sankey O. F., Soignard E., McMillan P. F., Phys. Rev. B, 2003, 67(9), 094104 |
| [35] | Liu Q. J., Ran Z., Liu F. S., Liu Z. T., J. Alloys Compd., 2015, 631, 192—201 |
| [36] | Soignard E., Somayazulu M., Dong J. J., Sankey O. F., McMillan P. F., J. Phys.: Condens. Matter., 2001, 13(4), 557—563 |
| [37] | Yu B. H., Chen D., Chin. Phys. B, 2012, 61(19), 197102 |
| [38] | Zhao Z. F., Li Y. F., Yao N., Chem. J. Chinese Universities, 2015, 36(8), 1648—1654 |
| (赵昨非, 李元丰, 姚宁. 高等学校化学学报, 2015, 36(8), 1648—1654) | |
| [39] | Petit A. T., Dulong P. L., Ann. Chim. Phys., 1819, 10, 395—413 |
| [1] | CHU Yuyi, LAN Chang, LUO Ergui, LIU Changpeng, GE Junjie, XING Wei. Single-atom Cerium Sites Designed for Durable Oxygen Reduction Reaction Catalyst with Weak Fenton Effect [J]. Chem. J. Chinese Universities, 2022, 43(9): 20220294. |
| [2] | ZHENG Anni, JIN Lei, YANG Jiaqiang, WANG Zhaoyun, LI Weiqing, YANG Fangzu, ZHAN Dongping, TIAN Zhongqun. Effects of 5,5-Dimethylhydantoin on Electroless Copper Plating [J]. Chem. J. Chinese Universities, 2022, 43(8): 20220191. |
| [3] | XIA Tian, WAN Jiawei, YU Ranbo. Progress of the Structure-property Correlation of Heteroatomic Coordination Structured Carbon-based Single-atom Electrocatalysts [J]. Chem. J. Chinese Universities, 2022, 43(5): 20220162. |
| [4] | WANG Hongning, HUANG Li, QING Jiang, MA Tengzhou, JIANG Wei, HUANG Weiqiu, CHEN Ruoyu. Activation of Biochar from Cattail and the VOCs Adsorption Application [J]. Chem. J. Chinese Universities, 2022, 43(4): 20210824. |
| [5] | LI Weihui, LI Haobo, ZENG Cheng, LIANG Haoyue, CHEN Jiajun, LI Junyong, LI Huiqiao. Hot-pressed PVDF-based Difunctional Protective Layer for Lithium Metal Anodes [J]. Chem. J. Chinese Universities, 2022, 43(2): 20210629. |
| [6] | CHANG Sihui, CHEN Tao, ZHAO Liming, QIU Yongjun. Thermal Degradation Mechanism of Bio-based Polybutylactam Plasticized by Ionic Liquids [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220353. |
| [7] | WANG Zumin, MENG Cheng, YU Ranbo. Doping Regulation in Transition Metal Phosphides for Hydrogen Evolution Catalysts [J]. Chem. J. Chinese Universities, 2022, 43(11): 20220544. |
| [8] | ZHUO Zengqing, PAN Feng. Progress of Key Electronic States in Lithium Ion Battery Materials Probed Through Soft X-ray Spectroscopy [J]. Chem. J. Chinese Universities, 2021, 42(8): 2332. |
| [9] | YUE Shengli, WU Guangbao, LI Xing, LI Kang, HUANG Gaosheng, TANG Yi, ZHOU Huiqiong. Research Progress of Quasi-two-dimensional Perovskite Solar Cells [J]. Chem. J. Chinese Universities, 2021, 42(6): 1648. |
| [10] | WANG Hongning, HUANG Li, SONG Fujiao, ZHU Ting, HUANG Weiqiu, ZHONG Jing, CHEN Ruoyu. Synthesis and VOCs Adsorption Properties of Hollow Carbon Nanospheres [J]. Chem. J. Chinese Universities, 2021, 42(6): 1704. |
| [11] | WANG Kunhua, YAO Jisong, YANG Junnan, SONG Yonghui, LIU Yuying, YAO Hongbin. Synthesis and Device Optimization of Highly Efficient Metal Halide Perovskite Light-emitting Diodes [J]. Chem. J. Chinese Universities, 2021, 42(5): 1464. |
| [12] | LIU Yao, DENG Zhengtao. Fast Synthesis of Highly Luminescent Two-dimensional Tin-halide Perovskites by Anti-solvent Method [J]. Chem. J. Chinese Universities, 2021, 42(12): 3774. |
| [13] | ZHANG Jun, WANG Bin, PAN Li, MA Zhe, LI Yuesheng. Synthesis and Properties of Imidazolium-based Polyethylene Ionomer [J]. Chem. J. Chinese Universities, 2020, 41(9): 2070. |
| [14] | WANG Tingting, LEI Yuhan, LIN Yujuan, HUANG Jialing, LIU Cuie, ZHENG Fengying, LI Shunxing. Preparation of Liposome-terminated CsPbX3(X=Cl,Br,I) Nanocrystals and Applications in Light-emitting Diode Devices [J]. Chem. J. Chinese Universities, 2020, 41(8): 1896. |
| [15] | SHI Haihan,WU Xiangping,PENG Xinzhe,YU Guojing,DONG Chaoyang,JI Yaoyao,YANG Siwen,CHEN Junlin,WANG Jin,RAN Xueqin,YANG Lei,XIE Linghai,HUANG Wei. An Effective Method of Reducing the Internal Reorganization Energy Based on Windmill-like Grid Composed of Four Carbazoles† [J]. Chem. J. Chinese Universities, 2020, 41(7): 1670. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||