

Chem. J. Chinese Universities ›› 2020, Vol. 41 ›› Issue (9): 2046.doi: 10.7503/cjcu20200291
• Physical Chemistry • Previous Articles Next Articles
SUN Tao, WANG Yibo( )
)
Received:2020-05-25
Online:2020-09-10
Published:2020-09-02
Contact:
WANG Yibo
E-mail:ybw@gzu.edu.cn
Supported by:CLC Number:
TrendMD:
SUN Tao, WANG Yibo. Theoretical Study on the Nature and Cooperation of C―H•••O and C―H•••π Interactions in the Pillar[5]arene and n⁃Alkanes Complexes[J]. Chem. J. Chinese Universities, 2020, 41(9): 2046.
| Method | ΔEb/(kJ·mol-1) | Method | ΔEb/(kJ·mol-1) | ||
|---|---|---|---|---|---|
| def2?SVPD | def2?TZVPP | def2?SVPD | def2?TZVPP | ||
| CCSD(T)/CBS | -23.77 | -23.77 | PBE0?D3BJ | -24.69 | -24.89 |
| ωB97X?V | -24.31 | -24.81 | B3LYP?D3BJ | -25.10 | -25.31 |
| ωB97M?V | -24.48 | -25.19 | B3PW91?D3BJ | -24.85 | -25.15 |
| ωB97X?D | -28.03 | -28.58 | CAM?B3LYP?D3BJ | -23.97 | -24.14 |
| M06?2X?D3 | -23.26 | -23.10 | LC?ωPBE?D3BJ | -24.56 | -24.77 |
| PBE?D3BJ | -24.94 | -25.15 | B2PLYP?D3BJ | -21.55 | -21.92 |
| Method | ΔEb/(kJ·mol-1) | Method | ΔEb/(kJ·mol-1) | ||
|---|---|---|---|---|---|
| def2?SVPD | def2?TZVPP | def2?SVPD | def2?TZVPP | ||
| CCSD(T)/CBS | -23.77 | -23.77 | PBE0?D3BJ | -24.69 | -24.89 |
| ωB97X?V | -24.31 | -24.81 | B3LYP?D3BJ | -25.10 | -25.31 |
| ωB97M?V | -24.48 | -25.19 | B3PW91?D3BJ | -24.85 | -25.15 |
| ωB97X?D | -28.03 | -28.58 | CAM?B3LYP?D3BJ | -23.97 | -24.14 |
| M06?2X?D3 | -23.26 | -23.10 | LC?ωPBE?D3BJ | -24.56 | -24.77 |
| PBE?D3BJ | -24.94 | -25.15 | B2PLYP?D3BJ | -21.55 | -21.92 |
| Complex | (kJ·mol-1) | (kJ·mol-1) | Complex | (kJ·mol-1) | (kJ·mol-1) |
|---|---|---|---|---|---|
| MeP5…CH4 | -32.68 | -30.33 | MeP5…C8H18 | -129.37 | -113.80 |
| MeP5…C2H6 | -55.69 | -49.62 | MeP5 …C9H20 | -133.43 | -117.53 |
| MeP5 …C3H8 | -74.35 | -65.73 | MeP5…C10H22 | -137.74 | -121.38 |
| MeP5…C4H10 | -91.42 | -81.34 | MeP5…C12H26 | -141.92 | -124.77 |
| MeP5…C5H12 | -103.85 | -91.17 | MeP5…C14H30 | -144.10 | -126.52 |
| MeP5…C6H14 | -112.97 | -100.29 | MeP5…C16H34 | -144.64 | -126.98 |
| MeP5…C7H16 | -121.63 | -106.82 |
| Complex | (kJ·mol-1) | (kJ·mol-1) | Complex | (kJ·mol-1) | (kJ·mol-1) |
|---|---|---|---|---|---|
| MeP5…CH4 | -32.68 | -30.33 | MeP5…C8H18 | -129.37 | -113.80 |
| MeP5…C2H6 | -55.69 | -49.62 | MeP5 …C9H20 | -133.43 | -117.53 |
| MeP5 …C3H8 | -74.35 | -65.73 | MeP5…C10H22 | -137.74 | -121.38 |
| MeP5…C4H10 | -91.42 | -81.34 | MeP5…C12H26 | -141.92 | -124.77 |
| MeP5…C5H12 | -103.85 | -91.17 | MeP5…C14H30 | -144.10 | -126.52 |
| MeP5…C6H14 | -112.97 | -100.29 | MeP5…C16H34 | -144.64 | -126.98 |
| MeP5…C7H16 | -121.63 | -106.82 |
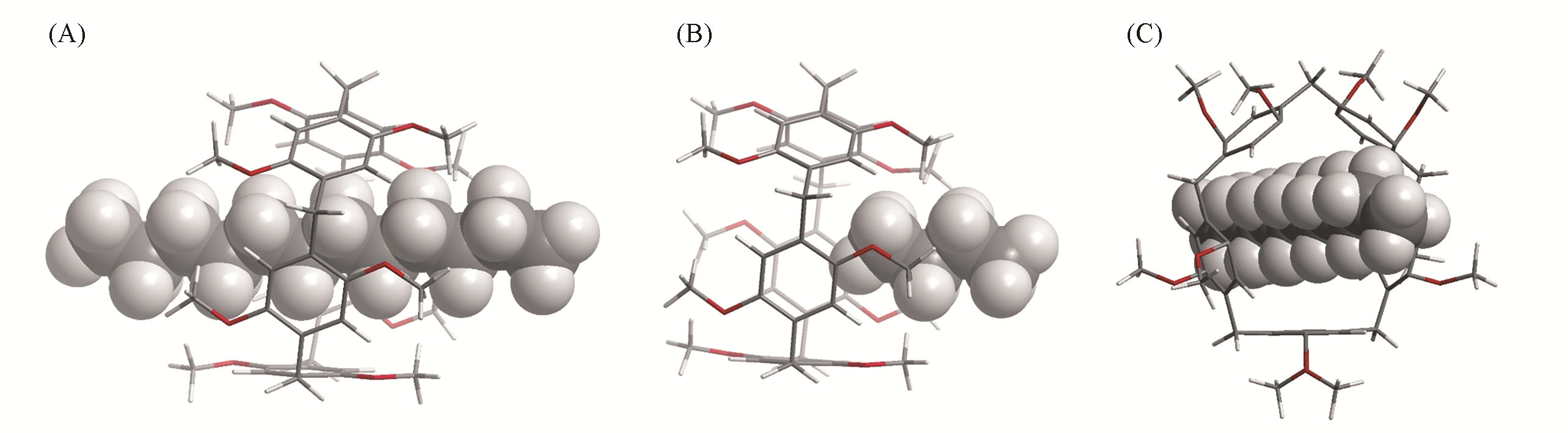
| Complex | EALMO-EDA/(kJ·mol-1) | EElec/(kJ·mol-1) | EPauli/(kJ·mol-1) | EDisp/(kJ·mol-1) | EPol/(kJ·mol-1) | ECT/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| MeP5…C12H26 | -124.75 | -210.48(46.06%) | 332.23 | -219.45(48.02%) | -15.28(3.34%) | -11.76(2.57%) |
| Model A | -21.18 | -72.98(47.92%) | 131.10 | -66.15(43.44%) | -5.13(3.37%) | -8.03(5.27%) |
| Model B | -72.25 | -136.60(47.98%) | 212.49 | -131.44(46.16%) | -8.26(2.90%) | -8.43(2.96%) |
| Complex | EALMO-EDA/(kJ·mol-1) | EElec/(kJ·mol-1) | EPauli/(kJ·mol-1) | EDisp/(kJ·mol-1) | EPol/(kJ·mol-1) | ECT/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| MeP5…C12H26 | -124.75 | -210.48(46.06%) | 332.23 | -219.45(48.02%) | -15.28(3.34%) | -11.76(2.57%) |
| Model A | -21.18 | -72.98(47.92%) | 131.10 | -66.15(43.44%) | -5.13(3.37%) | -8.03(5.27%) |
| Model B | -72.25 | -136.60(47.98%) | 212.49 | -131.44(46.16%) | -8.26(2.90%) | -8.43(2.96%) |
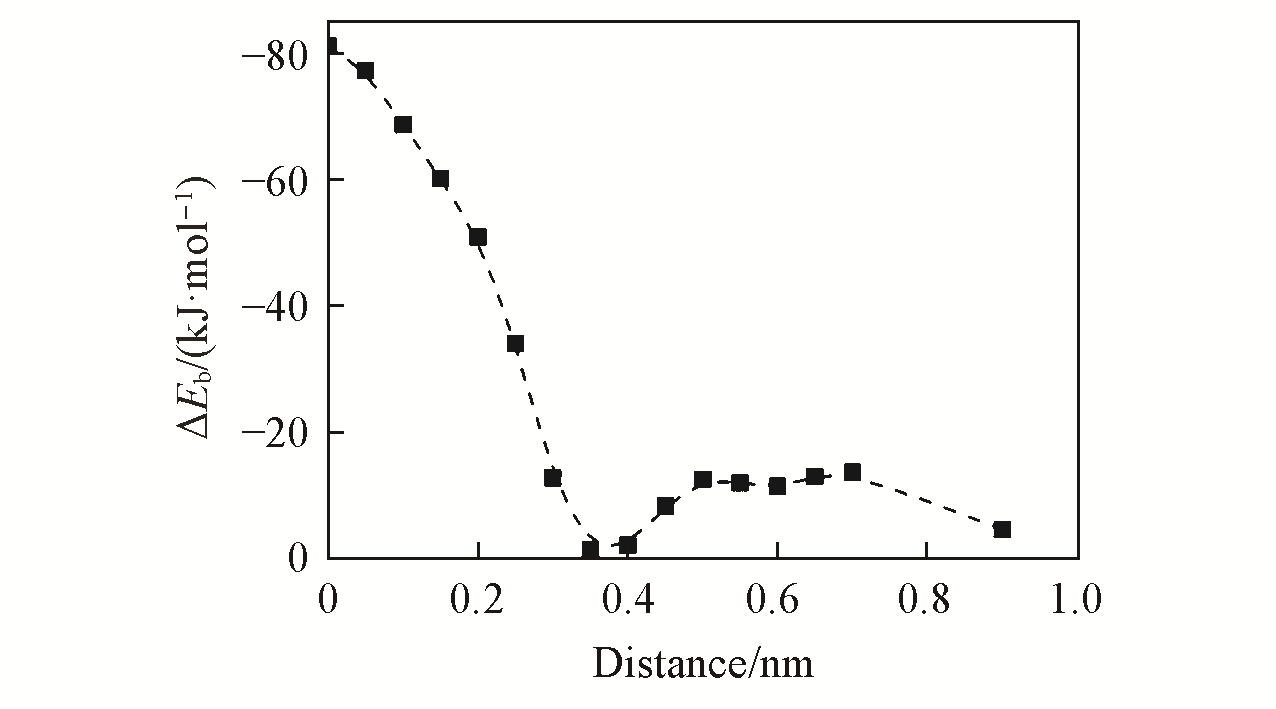
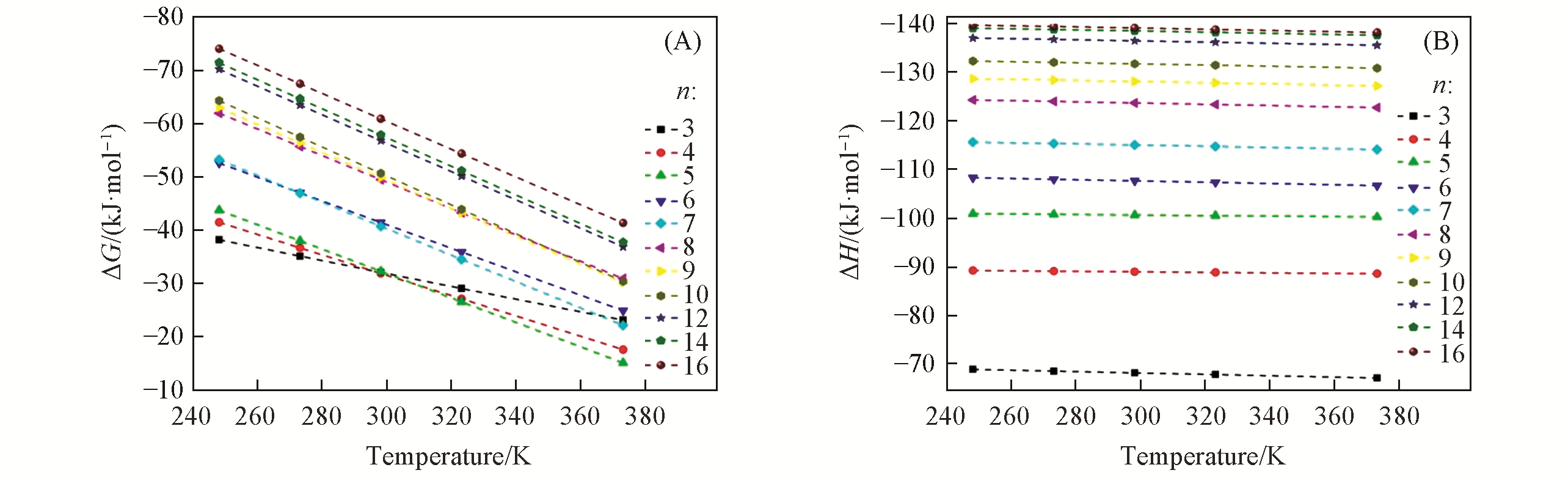
| Complex | EALMO-EDA/ (kJ·mol-1) | EElec/(kJ·mol-1) | EPauli/ (kJ·mol-1) | EDisp/(kJ·mol-1) | EPol/(kJ·mol-1) | ECT/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| MeP5…CH4 | -30.34 | -41.40(43.88%) | 64.01 | -47.75(50.61%) | -2.85(3.02%) | -2.35(2.49%) |
| MeP5…C2H6 | -49.62 | -80.48(46.13%) | 124.83 | -83.77(48.02%) | -5.79(3.32%) | -4.41(2.53%) |
| MeP5…C3H8 | -65.72 | -84.80(43.68%) | 128.43 | -99.12(51.06%) | -6.19(3.19%) | -4.04(2.08%) |
| MeP5…C4H10 | -81.34 | -107.92(43.70%) | 165.62 | -124.55(50.43%) | -8.51(3.44%) | -5.99(2.42%) |
| MeP5…C5H12 | -91.17 | -140.68(45.25%) | 219.71 | -150.93(48.55%) | -10.99(3.53%) | -8.28(2.66%) |
| MeP5 …C6H14 | -100.28 | -159.50(45.52%) | 250.15 | -168.81(48.17%) | -12.33(3.52%) | -9.79(2.79%) |
| MeP5…C7H16 | -106.80 | -181.70(46.40%) | 284.80 | -185.67(47.41%) | -13.49(3.45%) | -10.74(2.74%) |
| MeP5…C8H18 | -113.79 | -191.62(46.22%) | 300.76 | -197.79(47.71%) | -14.19(3.42%) | -10.95(2.64%) |
| MeP5…C9H20 | -117.48 | -199.39(46.23%) | 313.78 | -206.06(47.78%) | -14.59(3.38%) | -11.22(2.60%) |
| MeP5…C10H22 | -121.39 | -204.88(46.09%) | 323.10 | -213.30(47.99%) | -14.85(3.34%) | -11.46(2.58%) |
| MeP5…C12H26 | -124.75 | -210.48(46.06%) | 332.23 | -219.45(48.02%) | -15.28(3.34%) | -11.76(2.57%) |
| MeP5…C14H30 | -126.53 | -212.03(45.92%) | 335.19 | -222.26(48.14%) | -15.45(3.35%) | -11.98(2.60%) |
| MeP5…C16H34 | -126.96 | -212.72(45.92%) | 336.25 | -223.09(48.16%) | -15.50(3.35%) | -11.89(2.57%) |
| Complex | EALMO-EDA/ (kJ·mol-1) | EElec/(kJ·mol-1) | EPauli/ (kJ·mol-1) | EDisp/(kJ·mol-1) | EPol/(kJ·mol-1) | ECT/(kJ·mol-1) |
|---|---|---|---|---|---|---|
| MeP5…CH4 | -30.34 | -41.40(43.88%) | 64.01 | -47.75(50.61%) | -2.85(3.02%) | -2.35(2.49%) |
| MeP5…C2H6 | -49.62 | -80.48(46.13%) | 124.83 | -83.77(48.02%) | -5.79(3.32%) | -4.41(2.53%) |
| MeP5…C3H8 | -65.72 | -84.80(43.68%) | 128.43 | -99.12(51.06%) | -6.19(3.19%) | -4.04(2.08%) |
| MeP5…C4H10 | -81.34 | -107.92(43.70%) | 165.62 | -124.55(50.43%) | -8.51(3.44%) | -5.99(2.42%) |
| MeP5…C5H12 | -91.17 | -140.68(45.25%) | 219.71 | -150.93(48.55%) | -10.99(3.53%) | -8.28(2.66%) |
| MeP5 …C6H14 | -100.28 | -159.50(45.52%) | 250.15 | -168.81(48.17%) | -12.33(3.52%) | -9.79(2.79%) |
| MeP5…C7H16 | -106.80 | -181.70(46.40%) | 284.80 | -185.67(47.41%) | -13.49(3.45%) | -10.74(2.74%) |
| MeP5…C8H18 | -113.79 | -191.62(46.22%) | 300.76 | -197.79(47.71%) | -14.19(3.42%) | -10.95(2.64%) |
| MeP5…C9H20 | -117.48 | -199.39(46.23%) | 313.78 | -206.06(47.78%) | -14.59(3.38%) | -11.22(2.60%) |
| MeP5…C10H22 | -121.39 | -204.88(46.09%) | 323.10 | -213.30(47.99%) | -14.85(3.34%) | -11.46(2.58%) |
| MeP5…C12H26 | -124.75 | -210.48(46.06%) | 332.23 | -219.45(48.02%) | -15.28(3.34%) | -11.76(2.57%) |
| MeP5…C14H30 | -126.53 | -212.03(45.92%) | 335.19 | -222.26(48.14%) | -15.45(3.35%) | -11.98(2.60%) |
| MeP5…C16H34 | -126.96 | -212.72(45.92%) | 336.25 | -223.09(48.16%) | -15.50(3.35%) | -11.89(2.57%) |
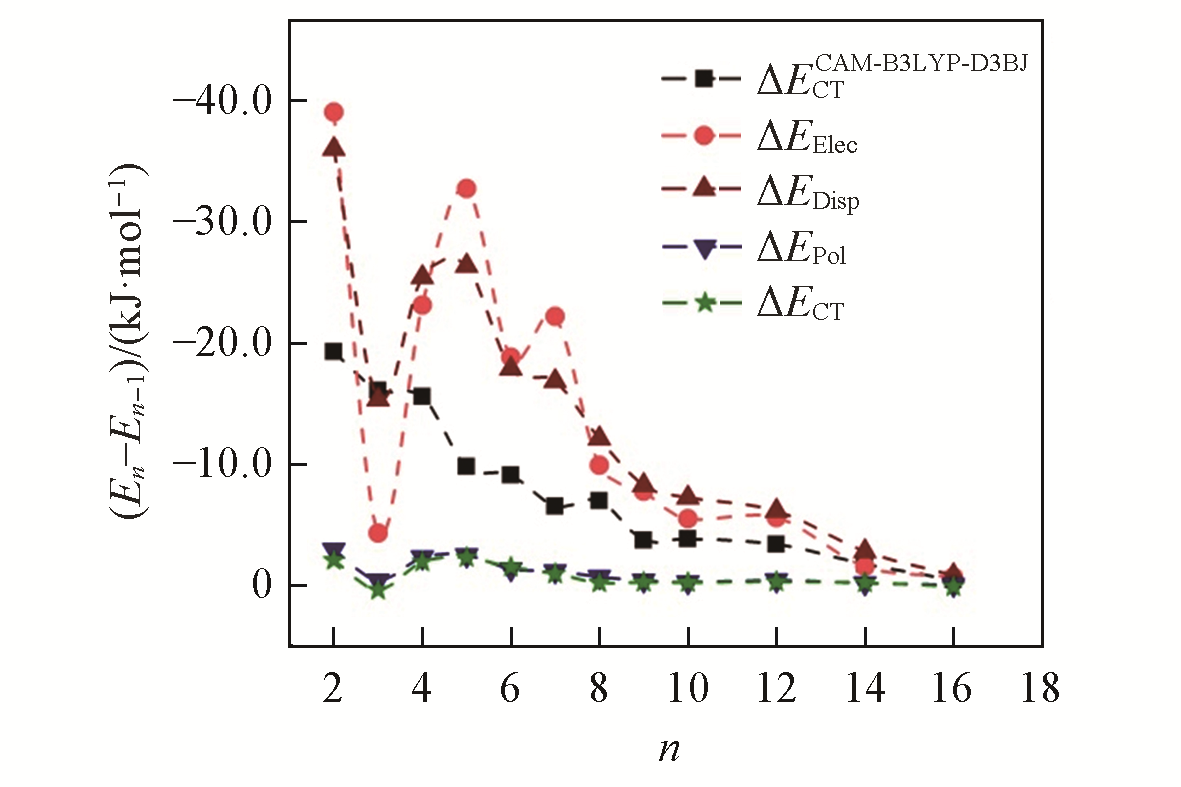
| 1 | Ogoshi T., Kanai S., Fujinami S., Yamagishi T. A., Nakamoto Y., J. Am. Chem. Soc., 2008, 130(15), 5022―5023 |
| 2 | Yu G., HanC., Zhang Z., Chen J., Yan X., Zheng B., Liu S., Huang F. H., J. Am. Chem. Soc., 2012, 134(20), 8711―8717 |
| 3 | Han C. Y., Zhang Z. B., Chi X. D., Zhang M. M., Yu G. C., Huang F. H., Acta Chim. Sinica, 2012, 70(17), 1775―1778(韩成友, 张子彬, 池小东, 张明明, 喻国灿, 黄飞鹤. 化学学报, 2012, 70(17), 1775―1778) |
| 4 | Dai D. H., Li Z., Wang C. Y., Wu J. R., Wang Y., Zhang D. M., Yang Y. W., J. Am. Chem. Soc.,2019, 141(11), 4756―4763 |
| 5 | Wang X. H., Song N., Hou W., Wang C. Y., Wang Y., Tang J., Yang Y. W., Adv. Mater., 2019, 31(37), 1903962 |
| 6 | Song N., Kakuta T., Yamagishi T., Yang Y. W., Ogoshi T., Chem.,2018,4(9), 2029―2053 |
| 7 | Wang K., Yang Y. W., Zhang X. A., Chem. J. Chinese Universities, 2012, 33(1), 1―13(王凯, 杨英威, 张晓安. 高等学校化学学报, 2012, 33(1), 1―13) |
| 8 | Tan L. L., Li H., Tao Y., Zhang S. X., Wang B., Yang Y. W., Adv. Mater.,2014, 26(41), 7027―7031 |
| 9 | Tan L. L., Zhu Y., Long H., Jin Y., Zhang W., Yang Y. W., Chem. Commun., 2017, 53(48), 6409―6412 |
| 10 | Zhang Z. B., Xia B. Y., Han C. Y., Yu Y. H., Huang F. H., Org. Lett., 2010,12(15), 3285―3287 |
| 11 | Zhang Z. B., Luo Y., Chen J. Z., Dong S. Y., Yu Y. H., Ma Z., Huang F. H., Angew. Chem. Int. Ed., 2011, 50(6), 1397―1401 |
| 12 | Zhang Z. B., Han C. Y., Yu G. C., Huang F. H., Chem. Sci., 2012, 3(10), 3026―3031 |
| 13 | Li C. J., Chen S. H., Li J., Han K., Xu M., Hu B. J., Yu Y. H., Jia X. S., Chem. Commun., 2011, 47(40), 11294―11296 |
| 14 | Shu X. Y., Chen S. H., Li J., Chen Z. X., Weng L. H., Jia X. S., Li C. Y., Chem. Commun., 2012, 48(24), 2967―2969 |
| 15 | Han K., Zhang Y. Y., Li J., Yu Y. H., Jia X. S., Li C. J., Eur. J. Org. Chem.,2013,2013(11), 2057―2060 |
| 16 | Ogoshi T., Sueto R., Yoshikoshi K., Yamaqishi T., Chem. Commun., 2014, 50(96), 15209―15211 |
| 17 | Ogoshi T., Demachi K., Kitajima K., Yamaqishi T., Chem. Commun., 2011,47(37), 10290―10292 |
| 18 | Ogoshi T., Sueto R., Yoshikoshi K., Sakata Y., Akine S., Yamagishi T., Angew. Chem. Int. Ed., 2015, 54(34), 9849―9852 |
| 19 | Ogoshi T., Sueto R., Hamada Y., Doitomi K., Hirao H., Sakata Y., Akine S., Kakuta T., Yamagishi T., Chem. Commun., 2017, 53(61),8577―8580 |
| 20 | Suvitha A., Venkataramanan N. S., J. Incl. Phenom. Macrocycl. Chem., 2017,87(1/2), 207―218 |
| 21 | Venkataramanan N. S., Suvitha A., Vijayaraghavan A., Thamotharan S., J. Mol. Liq., 2017, 241, 782―791 |
| 22 | Bhadane S. A., Lande D. N., Gejji S. P., J. Phys. Chem. A, 2016, 120(43), 8738―8749 |
| 23 | Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys., 2010, 132(15), 154104 |
| 24 | Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem., 2011, 32(7), 1456―1465 |
| 25 | Mardirossian N., Head⁃Gordon M., Phys. Chem. Chem. Phys., 2014,16(21), 9904―9924 |
| 26 | Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys., 2005, 7(18), 3297―3305 |
| 27 | Riley K. E., Pitoňák M., Jurečka P., Hobza P., Chem. Rev., 2010, 110(9), 5023―5063 |
| 28 | Chai J. D., Head⁃Gordon M., Phys. Chem. Chem. Phys., 2008, 10(44), 6615―6620 |
| 29 | Goerigk L., Grimme S., Phys. Chem. Chem. Phys., 2011, 13(14), 6670―6688 |
| 30 | Wang Y. P., Study on Quantum Chemistry Method to Solve the Thermodynamic Functions in the Intermolecular Interaction, Guizhou University, Guiyang, 2018(王裕平. 分子间相互作用热力学函数的量子化学精确计算方法研究, 贵阳: 贵州大学, 2018) |
| 31 | Boys S. F., Bernardi F., Mol. Phys., 1970,19(4), 553―566 |
| 32 | Horn P. R., Mao Y., Head⁃Gordon M., J. Chem. Phys., 2016, 144(11), 114107 |
| 33 | Horn P. R., MaoY., Head⁃Gordon M., Phys. Chem. Chem. Phys., 2016, 18(33), 23067—23079 |
| 34 | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams⁃Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J., Gaussian 16, Revision A. 03, Gaussian Inc., Wallingford CT., 2016 |
| 35 | Shao Y. H., Gan Z. T., Epifanovsky E., Gilbert A. T. B., Wormit M., Kussmann J., Lange A. W., Behn A., Deng J., Feng X. T., Ghosh D., Goldey M., Horn P. R., Jacobson L. D., Kaliman I., Khaliullin R. Z., Kus T., Landau A., Liu J., Proynov E. I., Rhee Y. M., Richard R. M., Rohrdanz M. A., Steele R. P., Sundstrom E. J., Woodcock H. L., Zimmerman P. M., Zuev D., Albrecht B., Alguire E., Austin B., Beran G. J. O., Bernard Y. A., Berquist E., Brandhorst K., Bravaya K. B., Brown S. T., Casanova D., Chang C. M., Chen Y. Q., Chien S. H., Closser K. D., Crittenden D. L., Diedenhofen M., DiStasio R. A., Do H., Dutoi A. D., Edgar R. G., Fatehi S., FustiMolnar L., Ghysels A., Golubeva⁃Zadorozhnaya A., Gomes J., Hanson⁃Heine M. W. D., Harbach P. H. P., Hauser A. W., Hohenstein E. G., Holden Z. C., Jagau T. C., Ji H. J., Kaduk B., Khistyaev K., Kim J., King R. A., Klunzinger P., Kosenkov D., Kowalczyk T., Krauter C. M., Lao K. U., Laurent A. D., Lawler K. V., Levchenko S. V., Lin C. Y., Liu F., Livshits E., Lochan R. C., Luenser A., Manohar P., Manzer S. F., Mao S. P., Mardirossian N., Marenich A. V., Maurer S. A., Mayhall N. J., Neuscamman E., Oana C. M., Olivares⁃Amaya R., O’Neill D. P., Parkhill J. A., Perrine T. M., Peverati R., Prociuk A., Rehn D. R., Rosta E., Russ N. J., Sharada S. M., Sharma S., Small D. W., Sodt A., Stein T., Mol. Phys., 2015, 113(2), 184—215 |
| 36 | Elm J., Passananti M., Kurtén T., Vehkamäki H., J. Phys. Chem. A, 2017,121(32), 6155—6164 |
| 37 | Witte J., Neaton J. B., Head-Gordon M., J. Chem. Phys., 2017, 146(23), 234105 |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||