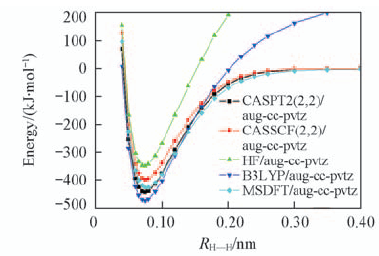
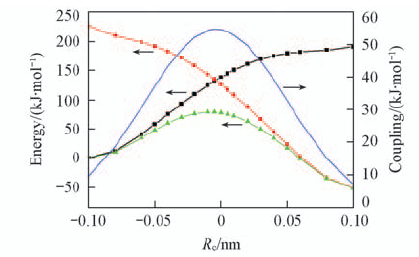
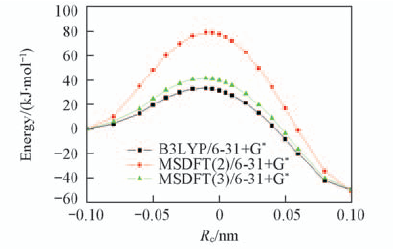
| [1] |
Subotnik J. E., Yeganeh S., Cave R. J., Ratner M. A., J. Chem. Phys., 2008, 129, 244101
|
| [2] |
Song L., Gao J., J. Phys. Chem. A, 2008, 112, 12925—12935
|
| [3] |
Mo Y., J. Chem. Phys., 2007, 126, 224104
|
| [4] |
Hoyer C. E., Xu X., Ma D., Gagliardi L., Truhlar D. G., J. Chem. Phys., 2014, 141, 114104
|
| [5] |
Shearer J., Schmitt J. C., Clewett H. S., J. Phys. Chem. B, 2015, 119, 5453—5461
|
| [6] |
Subotnic J. E., Alguire E. C., Ou Q., Landry B. R., Fatehi S., Acc. Chem. Res., 2015, 48, 1340—1350
|
| [7] |
Wu Q., vanVoorhis T., J. Chem. Phys., 2006, 125, 164105
|
| [8] |
Ye S., Geng C. Y., Shaik S., Neese F., Phys. Chem. Chem. Phys., 2013, 15, 8017—8030
|
| [9] |
Mo Y., Song L., Lin Y., Liu M., Cao Z., Wu W., J. Chem. Theory Comput., 2012, 8, 800—805
|
| [10] |
Chen H., Song J., Lai W., Wu W., Shaik S., J. Chem. Theory Comput., 2010, 6, 940—953
|
| [11] |
Steinmann S. N., Corminboeuf C., Wu W., Mo Y., J. Phys. Chem. A, 2011, 115, 5467—5477
|
| [12] |
Song L., Mo Y., Zhang Q., Wu W., J. Comput. Chem., 2005, 26, 514—521
|
| [13] |
Chen Z., Chen X., Wu W., J. Chem. Phys., 2013, 138, 164120
|
| [14] |
Chang X., Su P., Wu W., Chem. Phys. Lett., 2014, 610, 246—250
|
| [15] |
Wu W., Su P. F., Shaik S., Hiberty P. C., Chem. Rev., 2011, 111, 7557—7593
|
| [16] |
Song L., Mo Y., Gao J., J. Chem. Theory Comput., 2009, 5, 174—185
|
| [17] |
Gao J., Cembran A., Mo Y., J. Chem. Theory Comput., 2010, 6, 2402—2410
|
| [18] |
Song L., Gao J., J. Phys. Chem. A, 2008, 112, 12925—12935
|
| [19] |
Mo Y., Gao J., J. Phys. Chem. A, 2000, 104, 3012—3020
|
| [20] |
Cembran A., Provorse M. R., Wang C., Wei W., Gao J., J. Chem. Theory Comput., 2012, 8, 4347—4358
|
| [21] |
Cembarn A., Song L., Mo Y., Gao J., J. Chem. Theory Comput., 2009, 5, 2702—2716
|
| [22] |
Frisch M.J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2010
|
| [23] |
Werner H.J., Knowles P. J., Lindh R., Manby F. R., Schutz M., Celani P., Korona T., Rauhut G., Amos R. D., Bernhardsson A., et al., MOLPRO, Version 2010.1., A Package of ab initio Programs,
|
| [24] |
Schmidt M. W., Baldrdge K.K., Boatz J. A., Elbert S. T., Gordon M. S., Jensen J. H., Koseki S., Matsunaga N., Nguyen K. A., et al., J. Comput. Chem., 1993, 14, 1347—1363
|
| [25] |
Werner H. J., Mol. Phys., 1996, 89, 645—661
|
| [26] |
McCurdy P. R., Hess W. P. Xantheas S. S., J. Phys. Chem. A, 2002, 106, 7628—7653
|
| [27] |
Sedo G., Doran J. L., Leopold K. R., J. Phys. Chem. A, 2009, 113, 11301—11310
|
| [28] |
Bianco R., Wang S., Hynes J. T., J. Phys. Chem. A, 2007, 111, 11033—11042
|
| [29] |
Leopold K. R., Annu. Rev. Phys. Chem., 2011, 62, 327—349
|
| [30] |
Scott J. R., Wright J. B., J. Phys. Chem. A, 2004, 108, 10578—10585
|
| [31] |
D’Auria R., Turco R. P., Houk K. N., J. Phys. Chem. A, 2004, 108, 3756—3765
|
 )
)